When the disease occurs
The kidneys are formed, like all the internal organs of a child, inside the mother’s womb; up to six months they are noticeably different from the adult organ, and only by the first year of the child’s life do the kidneys completely return to normal.
Therefore, problems are sometimes identified in childhood, and sometimes only in adolescence, when the load on them increases significantly.
Diseases are preceded by congenital or acquired factors. Congenital causes include defects that appeared in the prenatal period:
- infectious, viral diseases of the mother during pregnancy;
- hereditary diseases;
- unhealthy lifestyle of a pregnant woman - smoking, drinking alcoholic beverages.
Acquired factors that can cause kidney disease in children:
- viral, bacterial infections;
- diabetes;
- poor diet, lack of water;
- weak immunity.
Frequent colds, sore throats, and tonsillitis can cause problems with the kidneys; improper hygiene of the genital organs causes inflammation in the genitourinary system, not forgetting to affect the kidneys. Girls are more likely to experience this than boys. Hypothermia reduces blood flow in all internal organs, including the kidneys, which causes inflammation.
HEREDITARY JADE-LIKE SYNDROMES
They are most often detected in the first two years of a child’s life on the basis of renal symptoms or in connection with abnormalities of other organs. Clinically, they manifest themselves with various symptoms and, in most cases, nonspecific. The latent period after acute infection cannot be established. In some syndromes, only hematuria or proteinuria is detected, in others - a picture of nephrotic syndrome, and in advanced cases - renal failure. Such atypia makes it difficult to distinguish between hereditary nephropathies from purchased. This distinction is essential for determining the treatment and prognosis of these diseases. In each individual case, the diagnosis is based not only on clinical signs, but also on a detailed study of family history. Genealogical analysis is one of the most significant research methods that allows us to detect the hereditary nature of the disease.
The presence of aminoaciduria is also of diagnostic importance. It is assumed that disruption of the tubular transport of sparingly soluble amino acids contributes to the development of hereditary nephropathies. In many of them, the interstitial metabolism of amino acids is disrupted not only in the kidneys, but also in other organs
Types of congenital diseases
The causes of congenital diseases are indicated above; some signs of kidney disease may already appear in newborns:
- enlarged tummy;
- boys have a weak stream of urine;
- change in urine color.
Yellowing of the skin in newborns, vomiting, and diarrhea are possible. If there is an inflammatory process in the kidneys, the baby’s body temperature rises. Therefore, if in the first days of life the baby notices a lack of urination or a restless state during this process, and even more so - convulsions, you should immediately contact a pediatrician.
Abnormalities of the kidney structure
Improper organ development in utero can cause problems with kidney function from birth.
Such anomalies include:
- doubling of the kidneys;
- duplication of the ureter;
- horseshoe-shaped organ.
Such kidney diseases in children are treated mainly with surgical intervention up to 1 year.
The occurrence of polycystic disease
This is a genetic disease in which fluid-filled cysts form and grow in the kidney tissue. Symptoms of the disease:
- bowel dysfunction;
- blood in urine;
- weakness of the body, fatigue, poor appetite;
- light-colored urine, excessive urination;
- skin itching;
- aching, nagging pain in the lower back.
Kidney function is not impaired, the pathology may not prevent a person from leading a normal lifestyle or may become chronic, in which case it is recommended to cleanse the kidneys by dialysis. If the child leads a healthy lifestyle, the disease is not fraught with serious consequences.
How does multicystic disease manifest?
A genetic or congenital pathology in which the renal tissue changes and the functions of the kidneys partially change requires medical supervision: if the cyst does not grow and does not put pressure on the organ, then surgical intervention may not be required. If the cyst grows, the organ will have to be removed.
Megaureter symptoms
Manifestations of the pathology are that urine from the bladder is thrown back into the urinary system.
The reasons are:
- poor development of the urethra;
- immaturity of the nervous system of a newborn baby.
It is usually eliminated surgically.
What is congenital hydronephrosis?
With this disease, urine stagnation occurs in the kidneys due to a narrowed or underdeveloped lumen of the lower part of the ureter. Usually, the pathology is detected before the birth of the baby during an ultrasound scan of a pregnant woman, then in the first months of life the pathology is eliminated surgically.
Congenital anomalies of the genitourinary system (CAMPS) account for 20–30% of all congenital anomalies detected by prenatal ultrasound. Around the world, about 40-50% of cases of renal failure in children are associated with congenital anomalies of the urinary system [1]. Congenital anomalies of the genitourinary system include a wide range of structural and functional diseases of the kidneys and urinary tract. These include the following diseases: renal hypoplasia/dysplasia, renal agenesis, cystic kidney disease, duplication, obstruction at the level of the ureteropelvic and vesicoureteral segments, vesicoureteral reflux, megaureter, hydronephrosis and posterior urethral valves. They can be isolated or part of a multiorgan syndrome [2, 3]. VAMPS can be a consequence of chromosomal diseases caused by genomic or structural disorders; they are not hereditary diseases, although they are a genetically determined pathology. Chromosomal diseases are characterized, as a rule, by multiple malformations of various organs.
The frequency of damage to the kidneys and urinary organs in a number of chromosomal diseases can be quite high. With trisomy of chromosome 21, the loss of part of the long arm of chromosome 18, the frequency of malformations of the urinary system organs exceeds the frequency of similar anomalies of the urinary system organs in the population. With trisomy syndrome of chromosome 9, cryptorchidism and micropenis are observed in boys; with Pattau syndrome, polycystic kidney disease is common; with Shereshevsky-Turner syndrome, congenital kidney defects are also common; and with Miller-Dicker syndrome, cases of renal aganesia are described.
There is evidence that congenital kidney diseases can be the result of a mutation in one gene (monogenic VAMPS). This factor is confirmed by the following provisions:
- VAMPS can be familial in nature [4];
- monogenic mouse models have produced VAMPS phenotypes;
- multiorgan monogenic human syndromes may include VAMPS phenotypes.
Recently, the hypothesis about the monogenic nature of VAMPS was confirmed by the discovery of more than 20 genes responsible for the development of VAMPS in humans [5–9]. Previously, only a few genes were known whose mutations cause VAMPS. Most of them are found in people with familial syndromes, including HNF1B (Renal Cysts and Diabetes Syndrome) [10], PAX2 (Renal Coloboma Syndrome) [11] and EYA1 (brachio-otto -renal syndrome – branchio-oto-renal syndrome) [12].
One of the first VAMPS, the genetic basis of which was studied, was polycystic kidney disease. This disease is caused by mutations in the PKD1 and PKD2 genes, which encode the proteins polycystin 1 and polycystin 2, respectively. Until recently, molecular genetic diagnosis of this disease was difficult, but now it is possible to diagnose 90% of cases of hereditary polycystic kidney disease, which is very important for predicting the course of the disease and, especially, in relation to donor kidney transplantation. Autosomal recessive polycystic kidney disease is characterized by the appearance of bilateral kidney cysts and can begin in utero. This disease develops immediately after birth or during adolescence, depending on the penetrance of compound heterozygotes for recessive mutations in the PKHD1 gene.
Recent studies have shown that VAMPS can be caused by mutations in several genes (Table 1) with an autosomal dominant or recessive mode of inheritance [10-12, 13-41].
Table 1. Genes – causes of isolated VAMPS and syndromes with a predominance of the VAMPS phenotype in humans
| Gene symbol | Kidney phenotype | Extrarenal phenotype | Het/Homd mouse model | Literary source | |
| A. Dominant VAMPS | |||||
| BMP4 | Renal hypoplasia | Microophthalmia | VAMPS | [13] | |
| EYA1 | Multiple kidney disease, renal aplasia | Deafness, ear abnormalities, bronchial cysts | VAMPS | VAMPS | [12] |
| GATA3 | Renal dysplasia | Hypoparathyroidism, cardiac abnormalities, immunodeficiency, deafness | No | [14, 15] | |
| HNF1B | Renal hypoplasia, solitary kidney, horseshoe kidney | Diabetes mellitus, hyperuricemia, hypomagnesemia | No | [10] | |
| KAL1 | Kidney agenesis | Micropenis, bilateral cryptorchidism, anosmia | [17] | ||
| PAX2 | PMR, renal hypoplasia | Hearing loss | VAMPS | VAMPS | [11,18] |
| RET | Kidney agenesis | No | VAMPS | [19, 20] | |
| ROBO2 | VUR, vesicoureteral junction defects | None | No | VAMPS | [21] |
| SALL1 | Renal hypodysplasia, renal agenesis | Anomalies of the limbs, eyes, anal canal | VAMPS | VAMPS | [22] |
| SIX1 | Renal hypodysplasia, PMR | Deafness, ear defects, bronchial cysts | No | VAMPS | [38] |
| SIX2 | Renal hypodysplasia | None | No | VAMPS | [13, 28] |
| SIX5 | Renal hypodysplasia, PMR | Deafness, ear defects, bronchial cysts | No | No | [24] |
| SOX17 | VUR, obstruction at the level of the vesicoureteral segment | No | No | [25] | |
| TNXB | PMR | Joint hypermobility | No | No | [26] |
| UPK3A | Renal hypodysplasia | Defects of the face and limbs | No | VAMPS | [27] |
| WNT4 | Renal hypodysplasia | Sex formation disorder, adrenal and lung dysplasia (SERKAL) | No | VAMPS | [29-31] |
| CHD1L | Renal hypodysplasia, VUR, obstruction at the level of the vesicoureteral segment | No | [32] | ||
| DSTYK | Renal hypodysplasia, obstruction at the level of the vesicoureteral segment | Epilepsy | [33] | ||
| MUC1 | Medullary cystic kidney disease type 1 | – | ? | [42] | |
| UMOD | Medullary cystic kidney disease type 2 | Hyperuricemia | VAMPS | [34] | |
| B. Recessive VAMPS | |||||
| ACE | Absence or incomplete differentiation of proximal tubules | Pulmonary hypoplasia (Potter sequence), cranial abnormalities | No | VAMPS | [35, 36,39] |
| AGT | Similar to ACE | Similar to ACE | No | VAMPS | [35, 36] |
| AGTR1 | Similar to ACE | Similar to ACE | No | WAMPS? | [35, 36] |
| REN | Similar to ACE | Similar to ACE | No | VAMPS | [35, 36] |
| FGF20 | Bilateral renal agenesis | No | No | VAMPS | [37] |
| TRAP1 | VUR, renal agenesis | VACTERL syndrome | [40] | ||
| FRAS1 | Renal agenia | Cryptophthalmos, nasal and pharyngeal abnormalities, mental retardation and syndactyly | No | VAMPS | [41] |
| FREM2 | Renal agenia | Cryptophthalmos, nasal and pharyngeal abnormalities, mental retardation and syndactyly | No | VAMPS | [41] |
The occurrence of VAMPS is associated with a disruption of normal nephrogenesis and can be caused by mutations in the genes responsible for this process.
In order to understand and identify the causes of VAMPS, it is necessary to analyze in detail the process of development of the genitourinary system. Kidney development can be divided into the following stages: the emergence of the ureteric bud, the mesenchymal-epithelial transition (MET), the morphogenesis of the renal network and the development of the nephron (includes the morphogenesis of the proximal and distal tubules and glomerulogenesis) [4, 16, 43-46] (Table 2). The molecular control of these developmental processes is regulated by a large number of genes and signaling pathways.
Table 2. Genes regulating the development of the kidneys and upper urinary tract
| Stages of kidney development | Genes in humans | Genes in mice | ||||||
| Formation of the ureteric bud | BMP4, EMX2 EYA1 FOXC1 FOXC2 | GDNF GFRA1 GREM1 HOXA 11 ISL1 | ITGA8 LHX1 LIM1 OSR1 PAX2 | RET ROBO2 HS2ST SALL SIX | SIX2 SLIT2 SPRY1 WT1 HOXC 11 | HOXD11 | Bmp4 Eya1 Gata3 | Pax2 Ret Robo2 |
| Penetration into mesenchyme | BMP4 BMP7 EYA1 | FGF20 LIM 1 TGFB2 | WNT4 WNT9B FGF8 | FGF9 OSR1 SIX2 | SMAD4 TCF 21 | Fgf20 Wnt4 | ||
| Morphogenesis of the collecting system | BMP4 BMP7 GPC 3 GREM1 POD1 PTEN PAX2 | FFGR1 FFGR2 HOXA11 HOXC11 RARA SPRY1 | AGT (angiotensin) AGTR (angiotensin receptor 1 and 2) HOXD 11 MET WNT11 | Agt Agtr | ||||
| Nephron formation and maturation | PAX2 UMOD WTQ | AGT JAG1 Agt receptor | NOTCH2 | Umod | ||||
Disruption at any of the developmental stages, as demonstrated in mouse models, can result in the clinical phenotype of VAMPS. Understanding the molecular mechanisms that control the formation of the genitourinary system has shifted attention in the study of these processes from classical anatomical theories to the modern cellular and genetic principle of understanding the etiology of VAMPS [47]. Kidney development begins with the formation of bilateral pronephros (NDs), which move laterally in the embryonic body. Near the lower extremities, a protrusion called the ureteric bud (UB) is formed from the nephrotic anlage. It penetrates the nearby metanephrotic mesenchyme (MM), elongates, and branches into the future collecting system of the kidney. The region of the ureteric bud (UB), which does not grow into the MM, gives rise to the ureter connecting the kidney to the bladder. Abnormalities of the ureteric bud, maturation and differentiation of all layers of the distal ureter are associated with the development of VAMPS.
Genetic studies in mice have identified key genes and regulatory mechanisms that may cause VAMPS [4].
GENETIC MECHANISMS OF THE APPEARANCE OF VAMPS AT THE STAGE OF FORMATION OF THE URETERAL RUDIGUM
I. Ichikawa et al. proposed the “bud” theory—the ureteric bud must emerge at a specific location from the pronephros and enter the center of the MM for normal kidney development [47]. If the bud appears more rostral or caudal, it incorrectly penetrates into the MM, which leads to hypoplasia or dysplasia of the kidney due to impaired morphology and nephrogenesis. The absence or several ureteric buds, respectively, can lead to agenesis (unilateral or bilateral) or duplication of the kidney. Rostral or caudal displacement of the ureteric bud can also lead to ectopic entry into the bladder and cause obstruction at the level of the vesicoureteral segment or cause VUR. The “ureteric bud theory” was confirmed in studies on mice with disorders in the Bmp4, Grem1, Gdnf, Ret, Foxc1/c2, Robo2, Slit2, Spry1 and Itga8 genes, which are involved in determining the location of the ureteral bud and its development, leading to the occurrence of VAMPS [6]. In humans, mutations in the RET gene were initially found to cause multiple endocrine neoplasia [48] and Hirschsprung's disease [49]. Mutations in this gene also lead to the formation of VAMPS in fetuses in the form of bilateral renal hypoplasia/agenesis [50]. In addition, the role of the RET gene as a cause of VAMPS is confirmed by the fact that many patients with Hirschsprung disease have concomitant, undiagnosed VAMPS [51]. The regulatory mechanism of the GDNF-RET signaling pathway plays a central role in the formation of the ureteric bud, therefore mutations in the genes of this signaling pathway can lead to the development of VAMPS. It turned out that mutations in the genes of transcription factors such as PAX2, EYA1 and SALL1 lead to the occurrence of VAMPS with syndrome-specific extracranial manifestations. PAX2 mutations were first described in patients with Renal Coloboma syndrome, including renal hypoplasia, optic nerve abnormalities, and deafness [10]. To date, 55 diseases caused by mutations of the PAX2 gene have been described in the world [18]. Mutations of the EYA1 gene lead to the occurrence of BOR syndrome (branchio-otorenal syndrome), characterized by anomalies in the development of the external ear, neck cysts, hearing impairment, and anomalies in the development of the kidneys (from hypoplasia to agenesis) [12]. Interestingly, mutations in the SIX1 and SIX5 genes were first discovered in patients with EYA1-negative BOR syndrome and are likely to represent a rarer cause of the disease [23, 24]. SALL1 mutations lead to Townes-Brocks syndrome, characterized by abnormalities of the kidneys, anal canal, ear and limbs [22]. BMP4 is expressed in mesenchymal cells surrounding the Wolffian duct and inhibits the GDNF-RET regulatory mechanism [39].
GENETIC MECHANISMS OF THE APPEARANCE OF VAMPS AT THE STAGE OF MESENCHYMAL-EPITHELIAL TRANSITION
Once the ureteric bud penetrates the MM, activation of metanephrogenic mesenchymal cells is induced. This stage begins with the polarization of the mesenchyme to produce nephron epithelial cells, a process called mesenchymal-epithelial transition (MET) [43, 44]. WNT proteins play a key role in this process, in particular the WNT9b and WNT4 proteins [43, 44]. In addition, recent studies in mice have shown that the signaling mechanism involving WNT proteins is partially regulated [52]. Mutations of the WNT4 or SIX2 genes have been identified in children with VAMPS [13, 31]. Another important role in the MET process is played by fibroblast growth factor [43, 44]. In addition, data have recently been presented on a mutation in the FGF20 gene, which led to bilateral renal agenesis in three fetuses in consanguineous families [37].
GENETIC MECHANISMS OF THE APPEARANCE OF VAMPS AT THE STAGE OF FORMATION OF THE KIDNEY COLLECTING SYSTEM
The next stage of kidney development is the morphogenesis of the collecting system. During this period, nephrons are formed at the end of the rudiments of the collecting system. One of the factors regulating this process is angiotensin 2, which activates angiotensin receptors type 1 and 2, which stimulates morphogenesis [53]. Accordingly, mutations in components of the renin-angiotensin system, such as: AGT (angiotensinogen), REN (renin), ACE (angiotensin-converting enzyme) and AGTR1 (angiotensin II receptor type 1), are associated with severe phenotypic manifestations of VAMPS in the form of renal tubular dysgenesis [35]. These rare autosomal recessive diseases are characterized by early fetal anuria leading to oligohydramnios. In experiments on mice, various components of the renin-angiotensin system were inactivated. It has been established that mice with the NULL genotype for the AGTR2 and AGT genes [54] have VAMPS; this phenotype does not develop in mice with the NULL genotype for the ACE and REN genes. These facts highlight possible differences between mouse models and human diseases.
GENETIC MECHANISMS OF THE APPEARANCE OF VAMPS AT THE STAGE OF NEPHRON FORMATION
While much attention has been paid to the early stages of kidney formation, much less is known about the genetic regulation of the nephron formation process [46]. The only gene identified in humans that regulates this process is the UMOD gene. The uromodulin gene, Uromodulin (UMOD), encodes the Tamm-Horsfall protein, one of the most abundant proteins in the human body [55]. UMOD mutations are the cause of various renal diseases: polycystic kidney disease type 2 (MCKD2), familial juvenile hyperuricemic nephropathy (FJHN) and glomerulocystic kidney disease (GCKD) [34]. All these diseases have an autosomal dominant pattern of inheritance.
GENETIC MECHANISMS OF THE APPEARANCE OF VAMPS AT THE STAGE OF FORMATION OF THE CONNECTION OF THE URETER AND BLADDER
After the pronephros penetrates the cloaca, the ureteric bud appears and penetrates into the MM, then the ureter separates from the common pronephros duct (CND, the most distal segment of the pronephros duct) and penetrates the bladder. This process is called “maturation of the distal ureter” and depends on CND apoptosis and is regulated by a signaling pathway involving retinoic acid, LAR-family protein tyrosine phosphate receptors, Ret and Dlg1 [56–58]. The ureter consists of several cellular layers: the urothelium, subepithelial ureteral mesenchyme (or stromal layer), smooth muscle layer, and adventitia. In the smooth muscle layer at the level of the pelvis there are pacemaker cells that cause unidirectional peristalsis of the ureter. Disturbances in the genes SHH, GLI3, TSHZ3, BMP4, UPK2, UPK3, BRG1, TBX18, genes of the reninangiotensin system family, DLG1, KIT and HCN3 expressed in these cells lead to the development of megaureter, hydronephrosis, PMR and/or obstruction [59, 60].
It is believed that mutations in the genes that cause VAMPS, based on studies done in mice, can be used for genetic testing in humans. However, screening of patients for individual genes that cause VAMP showed that, with the exception of the HNF1B and PAX2 genes, mutations in most genes tested occur in a small number of cases [61]. Geno-phenotypic correlations among different mutation carriers in the same family can be very complex; a mutation in one gene can cause different anomalies. Added to this is the fact that many VAMPS remain undiagnosed [62].
Currently, there are ready-made panels designed to test groups of genes using high-throughput next-generation sequencing for the diagnosis of kidney and urinary tract diseases. These include, for example:
- KidneySeq™: A Comprehensive Genetic Kidney Disease Panel, examines 170 different genes responsible for 75 genitourinary diseases;
- Ion AmpliSeq™Inherited Disease Panel target gene list, diseases such as polycystic kidney disease, brachiotorenal syndrome, Wilms tumor are assessed;
- Autosomal Dominant and Recessive Polycystic Kidney Disease NextGen Sequencing (NGS) Panel, Autosomal Dominant Polycystic Kidney Disease Sequencing Panel, Ashkenazi Jewish Carrier Multi-Gene Expanded Panel include the diagnosis of polycystic kidney disease;
- Branchiootorenal Syndrome Panel, OtoSeq Hearing Loss Deletion/Duplication Panel, OtoSeq Hearing Loss Panel include the diagnosis of brachiotorenal syndrome;
- Congenital Central Hypoventilation NGS Panel includes the diagnosis of renal agenesis.
PROSPECTS FOR STUDYING THE GENES THAT CAUSE VAMPS
Currently, much of the research is aimed at identifying new genes in which mutations may cause VAMPS, to improve genetic testing and increase understanding of the etiology of a wide range of VAMPS phenotypes.
Next-generation sequencing (NGS) has made it possible to screen patients with VAMPS to look for mutations in several genes [63]. This analysis, in combination with whole exome sequencing, allowed us to identify new genes in which mutations are a possible cause of VAMSP (DSTYK, TRAP1, TNXB) [64-66]. Dual serine/threonine and tyrosine kinase (DSTYK) – activates phosphorylation of ERK, which leads to activation of the FGF receptor. DSTYK is located near FGF receptors in the ureteric bud and MM. Mutations in DSTYK can disrupt the FGF signaling pathway and lead to the development of VAMPS [35]. TNF receptor associated protein (TRAP1) is a heat shock protein 90 associated with mitochondrial chaperones and mainly expressed in the proximal tubule and ascending limb of the loop of Henle. Mutations of the TRAP1 gene can cause apoptosis caused by reactive oxygen species and stimulate the activation of stress genes and autophagy, which will consequently affect kidney development [15]. Tenascin XB (TNXB) is expressed in the urothelial layer of the vesicoureteral junction. Its more active expression is noted in the basal cells of the urothelium in patients with VUR compared to controls [26]. Mutations of the TNXB gene can lead to disruption of the obturator mechanism in the area of the vesicoureteral junction and to the occurrence of VUR. Previously, it was believed that mutations in the FRAS1, FREM2, GRIP1, FREM1) genes are associated with Fraser syndrome, but it has now been proven that they can also cause isolated abnormalities of the genitourinary system [67].
CONCLUSION
Although in most cases it is not possible to identify the causative genes of VAMPS, aberrations in certain regions of the genome make it possible to map new genes in which mutations may cause VAMPS. There is no doubt that in the coming years, next-generation sequencing will identify new mutations and genes involved in the development of VAMPS in humans. Combining data on these new genes with mutational studies in mice will determine the precise mechanisms and shed light on the etiology of the entire spectrum of structural abnormalities of the genitourinary system.
The work was carried out within the framework of the State Assignment.
LITERATURE
1. Woolf AS, Price KL, Scambler PJ, Winyard PJ. Evolving concepts in human renal dysplasia. J Am Soc Nephrol 2004; 15(4):998–1007.
2. Australian and New Zealand Dialysis and Transplant Registry. Annual Paediatric Report. 2012. 3. North American Pediatric Renal Transplant Cooperative Study. Annual Report. Rockville. MD: The EMMES Corporation. 2008.
4. Bulum B, Ozcakar ZB, Ustuner E, Dusunceli E, Kavaz A, Duman D, et al. High frequency of kidney and urinary tract anomalies in asymptomatic first-degree relatives of patients with CAKUT. Pediatr Nephrol 2013; 28(11):2143–2147.
5. Yosypiv IV. Congenital anomalies of the kidney and urinary tract: a genetic disorder? Int J Nephrol 2012; Article ID 909083, 10 pages. (URL: http:/ /dx.doi.org/10.1155/2012/909083)/
6. Chen F. Genetic and developmental basis for urinary tract obstruction. Pediatr Nephrol 2009; 24 (9): 1621–1632.
7. Renkema KY, Winyard PJ, Skovorodkin IN, Levtchenko E, Hindryckx A, Jeanpierre C, et al, Novel perspectives for investigating congenital anomalies of the kidney an urinary tract (CAKUT) Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association. Euro Renal Ass 2011; 26:3843–3851.
8. Sanna-Cherchi S, Caridi G, Weng PL, Scolari F, Perfumo F, Gharavi AG, et al. Genetic approaches to human renal agenesis/hypoplasia and dysplasia. 22. Kohlhase J, Wischermann A, Reichenbach H, Froster U, Engel W. Mutations Pediatr Nephrol 2007; 22 (10): 1675–1684.
9. Weber S. Novel genetic aspects of congenital anomalies of kidney and urinary tract. Curr Opin Pediatr 2012; 24(2); 212–218.
10. Lindner TH, Njolstad PR, Horikawa Y, Bostad L, Bell GI, Sovik O. A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of he pseudo-POU domain of hepatocyte nuclear factor-1ß. Hum Mol Genet 1999; 8 (11): 2001–2008.
11. Sanyanusin P, Schimmenti LA, McNoe LA, Ward TA, Pierpont ME, Sullivan MJ, et al. Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat Genet 1995; 9 (4): 358–364.
12. Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, et al. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet 1997; 15 (2): 157–164.
13. Weber S, Taylor JC, Winyard P, Baker KF, Sullivan-Brown J, Schild R, et al.. SIX2 and BMP4 mutations associate with anomalous kidney development. J Am Soc Nephrol 2008; 9(5):891–903.
14. Pandolfi PP, Roth ME, Karis A, Leonard MW, Dzierzak E, Grosveld FG, et al. Targeted disruption of the GATA3 gene causes severe abnormalities in the nervous system and in fetal liver haematopoiesis. Nat Genet 1995; 11 (1): 40–44.
15. Van Esch H, Groenen P, Nesbit MA, Schuffenhauer S, Lichtner P, Vanderlinden G, et al.. GATA3 haplo-insufficiency causes human HDR syndrome. // Nature 2000; 406(6794):419–422.
16. Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, et al.. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. // Nat Genet 1997; 17 (4): 384–385.
17. Hardelin JP, Levilliers J, del Castillo I, Cohen-Salmon M, Legouis R, Blanchard S, et al. . X chromosome-linked Kallmann syndrome: stop mutations validate the candidate gene. Proc Natl Acad Sci USA 1992; 89(17):8190–8194.
18. Bower M, Salomon R, Allanson J, Antignac C, Benedicenti F, Benetti E, et al. Update of PAX2 mutations in renal coloboma syndrome and establishment of a locus-specific database. Hum Mutat 2012; 33(3):457–466.
19. Skinner MA, Safford SD, Reeves JG, Jackson ME, Freemerman AJ. Renal aplasia in humans is associated with RET mutations. Am J Hum Genet 2008; 82(2):344–351.
20. Yang Y, Houle AM, Letendre J, Richter A. RET Gly691Ser mutation is associated with primary vesicoureteral reflux in the French-Canadian population from Quebec. Hum Mutat 2008; 29 (5): 695–702.
21. Lu W, van Eerde AM, Fan X, Quintero-Rivera F, Kulkarni S, Ferguson H, et al. . Disruption of ROBO2 is associated with urinary tract anomalies and concerns the risk of vesicoureteral reflux. Am J Hum Genet 2007; 80(4):616–632.
22. Kohlhase J, Wischermann A, Reichenbach H, Froster U, Engel W. Mutations in the SALL1 putative transcription factor gene cause Townes-Brocks syndrome. Nat Genet 1998; 18(1):81–83.
23. Ruf RG, Xu PX, Silvius D, Otto EA, Beekmann F, Muerb UT, et al. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc Natl Acad Sci USA 2004; 101(21):8090–8095.
24. Hoskins BE, Cramer CH, Silvius D, Zou D, Raymond RM, Orten DJ, et al. Transcription factor SIX5 is mutated in patients with branchio-oto-renal syndrome. Am J Hum Genet 2007; 80(4):800–804.
25. Gimelli S, Caridi G, Beri S, McCracken K, Bocciardi R, Zordan P, et al. Mutations in SOX17 are associated with congenital anomalies of the kidney and the urinary tract. Hum Mutat 2010; 31(12):1352–1359.
26. Gbadegesin RA, Brophy PD, Adeyemo A, Hall G, Gupta IR, Hains D, et al. TNXB Mutations Can Cause Vesicoureteral Reflux. J Am Soc Nephrol 2013; 24 (8): 1313-1322.
27. Jenkins D, Bitner-Glindzicz M, Malcolm S, Hu CC, Allison J, Winyard PJ, et al. De novo Uroplakin IIIa heterozygous mutations cause human renal adysplasia leading to severe kidney failure. J Am Soc Nephrol 2005; 16(7):2141–2149.
28. Self M, Lagutin OV, Bowling B, Hendrix J, Cai Y, Dressler GR, et al. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. Embo J 2006; 25(21):5214–5228.
29. Biason-Lauber A, Konrad D, Navratil F, Schoenle EJ. A WNT4 mutation associated with Mullerian-duct regression and virilization in a 46,XX woman. N Engl J Med 2004; 351(8):792–798.
30. Mandel H, Shemer R, Borochowitz ZU, Okopnik M, Knopf C, Indelman M, et al. SERKAL syndrome: an autosomal-recessive disorder caused by a loss-offunction mutation in WNT4. Am J Hum Genet 2008; 82(1):39–47.
31. Vivante A, Mark-Danielli M, Davidovits M, Harari-Steinberg O, Omer D, Gnatek Y, et al. Renal hypodysplasia associates with a WNT4 variant that causes aberrant canonical WNT signaling. J Am Soc Nephrol 2013; 24 (4): 550–558.
32. Brockschmidt A, Chung B, Weber S, Fischer DC, Kolatsi-Joannou M, Christ L, et al. CHD1L: a new candidate gene for congenital anomalies of the kidneys and urinary tract (CAKUT). Nephrol Dial Transplant 2012; 27(6):2355–2364.
33. Sanna-Cherchi S, Sampogna RV, Papeta N, Burgess KE, Nees SN, Perry BJ, et al. Mutations in DSTYK and Dominant Urinary Tract Malformations. Semin Nephrol. 2009; 29 (4): 321–337. N Engl J Med 2013; 369(7):621-629.
34. Hart TC, Gorry MC, Hart PS, Woodard AS, Shihabi Z, Sandhu J, et al. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J Med Genet 2002; 39(12):882–892.
35. Gribouval O, Gonzales M, Neuhaus T, Aziza J, Bieth E, Laurent N, et al. Mutations in genes in the renin-angiotensin system are associated with autosomal recessive renal tubular dysgenesis. Nat Genet 2005; 37(9):964–968.
36. Gribouval O, Moriniere V, Pawtowski A, Arrondel C, Sallinen SL, Saloranta C, et al. Spectrum of mutations in the renin-angiotensin system genes in autosomal recessive renal tubular dysgenesis. Hum Mutat 2012; 33 (2): 316–326.
37. Barak H, Huh SH, Chen S, Jeanpierre C, Martinovic J, Parisot M, et al. FGF9 and FGF20 maintain the stemness of nephron progenitors in mice and man. Dev Cell 2012; 22(6):1191–1207.
38. Ruf RG, Berkman J, Wolf MT, Nurnberg P, Gattas M, Ruf EM, et al. A gene locus for branchio-otic syndrome maps to chromosome 14q21.3-q24.3. J Med Genet 2003; 40 (7): 515–519.
39. Esther CR, Marino EM, Howard TE, Machaud A, Corvol P, Capecchi MR, et al. The critical role of tissue angiotensin-converting enzyme as revealed by gene targeting in mice. J Clin Invest 1997; 99(10):2375–2385.
40. McGregor L, Makela V, Darling SM, Vrontou S, Chalepakis G, Roberts C, et al. . Fraser syndrome and mouse blebbed phenotype caused by mutations in FRAS1/Fras1 encoding a putative extracellular matrix protein. Nat Genet 2003; 34(2):203–208.
41. Jadeja S, Smyth I, Pitera JE, Taylor MS, van Haelst M, Bentley E, et al. Identification of a new gene mutated in Fraser syndrome and mouse myelencephalic blebs. Nat Genet 2005; 37(5):520–525.
42. Kirby A, Gnirke A, Jaffe DB, Barešová V, Pochet N, Blumenstiel B, et al. Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat Genet 2013; 45(3):299–303.
43. Dressler GR. Advances in early kidney specification, development and patterning. Development 2009; 136(23):3863–3874.
44. Reidy KJ, Rosenblum ND. Cell and molecular biology of kidney development. Semin Nephrol 2009; 29 (4): 321–337.
45. Faa G, Gerosa C, Fanni D, Monga G, Zaffanello M, Van Eyken P, et al. Morphogenesis and molecular mechanisms involved in human kidney development. J Cell Physiol 2012; 227(3):1257–1268.
46. Vainio S, Lin Y. Coordinating early kidney development: lessons from gene targeting. Nat Rev Genet 2002; 3(7):533–543.
47. Ichikawa I, Kuwayama F, Pope JC, Stephens FD, Miyazaki Y. Paradigm shift from classic anatomic theories to contemporary cell biological views of CAKUT. Kidney Int 2002; 61(3):889–898.
48. Santoro M, Carlomagno F, Romano A, Bottaro DP, Dathan NA, Grieco M, et al. Activation of RET as a dominant transforming gene by germline mutations of MEN2A and MEN2B. Science 1995; 267 (5196):381–383.
49. Romeo G, Ronchetto P, Luo Y, Barone V, Seri M, Ceccherini I, et al. Point mutations affecting the tyrosine kinase domain of the RET proto-oncogene in Hirschsprung's disease. Nature 1994; 367(6461):377–378.
50. Jeanpierre C, Mace G, Parisot M, Moriniere V, Pawtowsky A, Benabou M, et al. RET and GDNF mutations are rare in fetuses with renal agenesis or other severe kidney development defects. J Med Genet 2011; 48(7):497–504.
51. Pini Prato A, Musso M, Ceccherini I, Mattioli G, Giunta C, Ghiggeri GM, et al. Hirschsprung disease and congenital anomalies of the kidney and urinary tract (CAKUT): a novel syndromic association. Medicine 2009; 88(2):83–90.
52. Park JS, Ma W, O'Brien LL, Chung E, Guo JJ, Cheng JG, et al. Six2 and Wnt regulate self-renewal and commitment of nephron progenitors through shared gene regulatory networks. Dev Cell 2012; 23 (3): pp. 637–651.
53. Reidy KJ, Rosenblum ND. Cell and molecular biology of kidney development. Semin Nephrol. 2009; 29 (4): 321–337.
54. Nishimura H, Yerkes E, Hohenfellner K, Miyazaki Y, Ma J, Hunley TE, Yoshida H, et al. Role of the angiotensin type 2 receptor gene in congenital anomalies of the kidney and urinary tract, CAKUT, of mice and men. Mol Cell 1999; 3(1):1–10.
55. Tamm I, Horsfall FLJr. Characterization and separation of an inhibitor of viral hemagglutination present in urine. Proc Soc Exp Biol Med 1950; 74 (1): 106–108.
56. Chia I, Grote D, Marcotte M, Batourina E, Mendelsohn C, Bouchard M. Nephric duct insertion is a crucial step in urinary tract maturation that is regulated by a Gata3-Raldh2-Ret molecular network in mice. Development 2011; 138(10):2089–2097.
57. Mackie GG, Stephens FD. Duplex kidneys: A correlation of renal dysplasia with position of the ureteral orifice. J Urol 1975; 114(2):274–280.
58. Costantini F. Genetic controls and cellular behaviors in branching morphogenesis of the renal collecting system. Wiley Interdiscip Rev Dev Biol 2012; 1(5):693–713.
59. Batourina E, Tsai S, Lambert S, Sprenkle P, Viana R, Dutta S, et al. Apoptosis induced by vitamin A signaling is crucial for connecting the ureters to the bladder. Nat Genet 2005; 37(10):1082–1089.
60. Uetani N, Bertozzi K, Chagnon MJ, Hendriks W, Tremblay ML, Bouchard M. Maturation of ureter-bladder connection in mice is controlled by LAR family receptor protein tyrosine phosphatases. J Clin Invest 2009; 119(4):924–935.
61. Kim ST, Ahn SY, Swat W, Miner JH. DLG1 influences distal ureter maturation via a non-epithelial cell autonomous mechanism involving reduced retinoic acid signaling, Ret expression, and apoptosis. Dev Biol. 2014; 390(2):160–169.
62. Bohnenpoll T, Kispert A. Ureter growth and differentiation. Semin Cell Dev Biol 2014; 36: 21–30.
63. Rasouly HM, Lu W. Lower urinary tract development and disease. Wiley Interdiscip Rev Syst Biol Med 2013; 5 (3): 307–342.
64. Renkema KY, Winyard PJ, Skovorodkin IN, Levtchenko E, Hindryckx A, Jeanpierre C, et al. Novel perspectives for investigating congenital anomalies of the kidney and urinary tract (CAKUT). Nephrol Dial Transplant 2011; 26(12):3843–3851.
65. Saisawat P, Tasic V, Vega-Warner V, Kehinde EO, Günther B, Airik R, et al. Identification of two novel CAKUT-causing genes by massively parallel exon resequencing of candidate genes in patients with unilateral renal agenesis. Kidney Int 2012; 81(2):196–200.
66. Saisawat P, Kohl S, Hilger AC, Hwang DY, Yung Gee H, Dworschak GC, et al. Whole-exome resequencing reveals recessive mutations in TRAP1 in individuals with CAKUT and VACTERL association. Kidney Int 2014; 85(6):1310–1317.
67. Kohl S, Hwang DY, Dworschak GC, Hilger AC, Saisawat P, Vivante A, et al. Mild recessive mutations in six Fraser syndrome-related genes cause isolated congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol 2014; 25 (9): 1917–1922.
| Attached file | Size |
| 595.68 kb |
‹ Fournier's gangrene: modern approaches to treatment (literature review) Up Clinical and economic rationale for the strategy of determining PCA3 in urine in the diagnosis of prostate cancer ›
Features of the course of glomerulonephritis
Acute, subacute, chronic glomerulonephritis can be a consequence of past inflammation:
- streptococcal infections;
- tonsillitis;
- faces;
- scarlet fever.
Complications can appear within just a few days: the child becomes lethargic, complains of headaches and discomfort in the abdomen.
Often observed:
- vomit;
- nausea;
- the face swells, then the legs;
- urine turns red.
The baby is prescribed a diet - foods without salt, limited fluid intake, but more vegetables and flour products. Antibacterial therapy is carried out and vitamins are prescribed.
Treatment must be started as soon as possible, otherwise there is a risk of acute renal failure, in which the kidneys fail and the body becomes intoxicated.
From the acute form, a subacute form can develop, characterized by the rapid development of chronic renal failure. It is necessary to urgently start therapy, otherwise the prognosis is unfavorable. Hemodialysis, diet and the same therapy as for the acute form can help.
Chronic glomerulonephritis is more common in children under five years of age in the nephrotic form.
Signs of kidney disease are:
- swelling;
- bloody urine;
- decreased protein levels in the blood;
- increased protein levels in the urine;
- blanching of the skin, cooling;
- dry mouth;
- shortness of breath, cough.
Periods of remission alternate with periods of exacerbation: symptoms subside and then appear again.
Treatment and symptoms of kidney inflammation in women
March 21, 2020
Inflammation of the kidneys (nephritis) is quite common in urology. Various factors can provoke pathology into activity, but as soon as symptoms of kidney inflammation in women begin to appear, it is necessary to urgently contact a medical facility. Nephritis is presented as an inflammatory process of the kidneys, which rapidly affects the glomerular apparatus in a woman’s body.
The inflammatory process is divided into 2 subtypes: focal (activated in several areas of the internal organ) and diffuse (infectious-inflammatory pathology affects both kidneys). Doctors note that diffuse inflammation of the kidneys poses a great danger to the female body. In this case, the inflammatory process affects all tissues of the internal organ, which, if adequate treatment is not followed, can lead to various complications.
Classification
Medical statistics indicate that the female half of the population is more susceptible to kidney inflammation. The root causes of pathology may be different, but the inflammatory process itself is classified into the following types:
- Nephritis in the interstitial form - the tubules of the internal organ are affected.
- Shunt nephritis - the inflammatory process affects the immune system.
- Pyelonephritis - inflammation develops due to the activity of harmful bacteria.
- Glomerulonephritis - inflammation develops due to damage to the internal organ of glomerular viruses.
Causes of kidney inflammation in women
As a rule, a pathological infection in a woman’s body spreads from the lower urinary tract. Pathogenic bacteria enter the body through the urethra (discomfort when urinating), which removes urine, and then multiply and affect the bladder, and then the kidneys. Bacteria from the bladder “move” into the kidneys through the ureters.
Inflammation can also be caused by pathogenic bacteria (Escherichia coli), which, when active, quickly damage the kidneys. If a woman has an infection in her body, it can spread to the kidneys through the blood and cause acute inflammation.
Main reasons:
- Congenital pathology of the development of internal organs - urethra, urinary canal, bladder;
- Urolithiasis disease;
- Pathologies of the urine outflow process;
- Elderly age;
- Weakness of the immune system due to diseases such as AIDS, malignant neoplasms, diabetes mellitus.
Urinary tract infections can also occur for reasons such as:
- Due to certain medical procedures (when a catheter is prescribed);
- After undergoing cystoscopy;
- After urinary tract surgery;
- After a course of medication;
- In case of spinal cord injury.
Secondary inflammation develops for the following reasons:
- Metal poisoning;
- The presence of serious pathologies in the body - tuberculosis, arthritis, diabetes, syphilis;
- Abuse of alcoholic beverages for a long time;
- Lesions of the skin and internal organs by benign or malignant neoplasms;
- Pregnancy;
- Flu, which leads to severe consequences;
- Chronic kidney diseases.
Signs of inflammation
When the inflammatory process begins, the symptoms of the pathology appear immediately. If your general health worsens, it is important to immediately contact a medical facility. Self-medication is very dangerous; it can negatively affect both the general health and the life of a woman.
What are the main symptoms?
- A sharp increase in temperature to a limit of 38 degrees or more;
- The appearance of sharp and nagging pain in the lumbar region, back, groin area and lower abdomen;
- Pain when urinating;
- Sedimentary phenomena appear in the urine in the form of flakes, and often inclusions of blood or pus are formed in the urine;
- “False” urge to urinate;
- The appearance of a “rotten” smell in the urine.
Secondary signs:
- Increased sweating;
- chills or fever;
- Nausea, often vomiting;
- General weakness, fatigue;
- "Sticky" skin;
- Clouding of consciousness (in older people);
- Swelling of the lower extremities, face;
- Violation of the normal functioning of the digestive tract (flatulence, diarrhea or constipation);
- Thirst;
- Feeling of dry mouth.
If symptoms and causes are ignored, the pathology provokes the development of complications such as the appearance of seizures, numbness of the skin, and muscle pain. Severe consequences of the inflammatory process of the kidneys are the development of hydrothorax or hydropericardium.
Basis of treatment
Treatment should begin immediately after the initial painful symptoms appear. At an early stage, the pathology can be cured quickly and without the use of strong medications. In its advanced form, the pathology can develop concomitant diseases, in which case the patient will require intensive therapy.
To choose the right treatment regimen, it is important to determine the type and determine the root cause of the pathology.
The first thing that is used as treatment is to eliminate the cause that provokes inflammation of the internal organ. If a woman is in serious condition, treatment is carried out in a hospital. In other cases, therapy at home is allowed, but with strict adherence to medical requirements.
The drug treatment regimen includes the following medications:
- Diuretics;
- Antibiotics;
- Antihypertensive drugs;
- Calcium preparations;
- Medicines that stabilize blood circulation, as well as nutrition in the renal tissues;
- Cytostatics;
- Antihistamines;
- Immunomodulators;
- Glucocorticosteroids;
- Vitamins of the B subgroup, as well as vitamins P and C.
All medications are prescribed by the doctor on an individual basis.
Therapeutic diet
Following a therapeutic diet during therapy will help eliminate the disease quickly and without negative consequences. The basis of the diet is to reduce the load on the internal affected organ. The patient's diet should include fresh vegetables, as well as fruits, cereals, and bran bread. Nutritionists recommend strictly limiting the consumption of meat and dairy products, as well as salt.
If a woman suffers from hypertension, she is recommended to drink less fluid. Otherwise, it is important to comply with the required daily drinking regimen (2 liters). Instead of drinking water, you can drink decoctions of medicinal herbs (birch buds, horsetail, juniper, corn silk, bearberry, sage) or fruit drinks from cranberries or lingonberries; such drinks help relieve the inflammatory process in the body and have an active diuretic property.
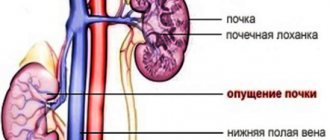
Causes of pyelonephritis
Pyelonephritis - acute or chronic - is called inflammation of the renal pelvis. Inflammation of the child’s kidneys occurs due to infection of the kidneys by pathogenic bacteria - staphylococci, E. coli, Proteus. Bacteria penetrate the kidneys through the blood (typical for infants from the first days of life), through the urinary tract or lymphatic vessels.
The genitourinary tract is typical for older children, and girls suffer from pyelonephritis more often than boys, which is due to the difference in the structure of the genitourinary system - girls have a shorter urethra than boys, so bacteria quickly penetrate through it into the kidneys.
Kidney inflammation is most severe in infants. Observed:
- high – up to 400 – body temperature;
- vomiting - intoxication is evident;
- the baby's legs bend, the head tilts back;
- the baby's skin turns yellow;
- dehydration occurs;
- The baby is restless - urination causes severe pain.
Older children experience a frequent urge to urinate, a burning sensation during this process, and pain in the lower back.
Treatment involves rest, preferably bed rest, a salt-free diet, taking antibiotics, and drinking plenty of fluids.
In chronic pyelonephritis, both kidneys are usually affected; during an exacerbation, the following are observed:
- pain and increased frequency of urination;
- feverish conditions;
- pain radiating to the lower back;
- severe headaches.
Treatment is usually prescribed the same as for acute pyelonephritis.
SHAFER SYNDROME
Etiology and pathogenesis. This syndrome is very similar to familial nephropathy with deafness (Alport syndrome). It was described by Schafer in 1962. Affected individuals have a high level of proline in the blood, and in some cases an enzyme defect—proline oxidase deficiency in the liver. The syndrome is inherited in an autosomal recessive manner.
Clinic. One of the main signs of the disease is microhematuria. Deafness of nervous origin is observed in some patients. Some children have seizures provoked by light. In such cases, electroencephalography shows increased sensitivity to light. Another important symptom is mental retardation - isolated or in combination with other signs
There is still no data on effective treatment. The prognosis is unfavorable.
Features of infectious nephrotic syndrome
The pathology is a toxic kidney injury, caused by intrauterine infections and birth asphyxia.
The disease begins:
- decreased appetite in a child;
- he becomes lethargic, irritable;
- the skin turns pale;
- swelling appears;
- there is an unpleasant aftertaste, dry mouth;
- the child feels sick, vomits;
- bloating and diarrhea are observed.
Treatment should be started as soon as possible - the doctor will prescribe the correct therapy and diet - diuretics, vitamins, Prednisolone and other drugs will speed up recovery.
HEREDITARY NEPHROSIS
Hereditary nephrosis is a rare disease that occurs soon after the birth of a child or in infancy. The disease is hereditary in nature; the prognosis is poor
Etiology and pathogenesis. The disease was first described in 1942, but the most detailed study is by Hallman. It is believed that the disease is inherited in an autosomal recessive manner. In approximately one third of families with this disease, consanguineous marriages have been established. The number of affected children of the same kind can reach 3-4. There are also cases of the disease in eight children - four pairs of homozygous twins. Usually the mother does not suffer from nephropathy, but pregnancy often occurs with toxicosis. Most of the reported cases of disease are from Scandinavian countries, mainly from Finland (Hallman). It is assumed that hereditary nephrosis is associated either with a congenital malformation of the kidneys or with a congenital disorder of protein synthesis. Hallman discovered the presence of anti-renal antibodies in the blood of a sick newborn child and in his mother. There is a possibility of immunization of the mother with kidney proteins and the transfer of antibodies through the placenta into the blood of the fetus, which leads to its disease even during intrauterine development. Blood group incompatibility (Rh factor) is also allowed as etiological factors.
The pathodotoanatomical picture of the disease is both typical and atypical. In many cases, changes in the kidneys are characterized by small kidney cysts
Clinic. Hereditary nephrosis most often develops in a newborn child or in the first months of life, in rare cases - until the end of the second year of life. In most cases, children are born premature, the placenta is edematous and large in size. In children, secondary changes in the bones appear early - wide sutures of the skull, a slowdown in the ossification process. The child's body weight and height may be significantly below normal.
Typical signs are swelling, general lethargy and pale skin. Significant changes occur in whey proteins, low levels of total protein, high hypoalbuminemia. The levels of total lipids and cholesterol in the blood serum are elevated. Proteinuria is significant. The diagnosis is aided by the results of a kidney biopsy.
Hereditary nephrosis is characterized by rapid progressive development. Many children die in the first few months of the first year of life
Treatment. Treatment with corticosteroids carried out to this day has not yielded results, with the exception of patients with minimal kidney damage. Diuretic drugs and a suitable protein-rich diet are indicated. If congenital nephrosis appears in the family, it is recommended to seek genetic counseling to avoid recurrence of the disease.