Adrenal myelolipoma is a fairly rare disease characterized by the presence of a hormonally inactive tumor consisting of adipose tissue and elements of the hematopoietic process. Such a formation is noted to be benign, and it is not difficult to detect it using ultrasound, cytological examination, computed tomography or magnetic resonance imaging.
What is and danger
Myelolipoma does not affect the production of hormones and is characterized by slow growth. May cause compression of the kidney and other structures of the system.
Need advice from an experienced doctor?
Get a doctor's consultation online. Ask your question right now.
Ask a free question
In most cases, myelolipoma develops in only one adrenal gland. Diagnosed in only 1% of patients with wen. It occurs in men and women aged 50 to 70 years.
A small tumor does not pose a threat to human life. If large in size, it can compress neighboring organs. In this case, the blood supply is disrupted and necrosis develops. If the diameter of the myelolipoma exceeds 10 cm, spontaneous rupture of the capsule is possible. In this case, adipose tissue and elements of the red brain enter the retroperitoneal space, and internal bleeding develops. This is a life-threatening condition.
According to ICD-10, the disease is assigned code D35.0 - benign formations of the endocrine glands.
Causes of the disease
Adrenal myelolipoma is a fairly rare disease. In modern medicine, during clinical studies, there is no accurate information explaining the causes of tumor development.
There are suspected factors that cause this formation:
- Genetic predisposition.
- Long-term intoxication of the body.
- Consequences of radiation therapy.
- Extensive burns leading to the formation of sepsis, which allows pathogenic microbes to enter the blood and cause infection of the body.
- Stress and poor quality nutrition.
- Uncontrolled division of bone marrow cells.
Causes and symptoms of pathology
The reasons for the development of the pathological process have not been determined. In people with obesity and endocrine disorders, such pathologies are more common than in other categories of patients.
There are several theories about the causes of myelolipoma:
- Failure during embryonic development. Pieces of red brain remained in the organ tissue. Hormonal imbalance subsequently created conditions for the formation of lipoma.
- Transfer of intravascular bone marrow substrate to the adrenal gland for unknown reasons.
- Violation of the function of cells of the hematopoietic system due to bacterial infection, treatment of cancer by surgery, aggressive drugs, after extensive thermal or chemical burns.
- Chronic stress, overwork combined with a violation of the principles of healthy eating.
When the size of the formation is small, there are no symptoms. Myelolipoma of the left or right adrenal gland is discovered by chance during a routine, preventive examination, ultrasound of the kidney, prescribed when examining a patient by a urologist or nephrologist.
With extensive tumors, it begins to compress neighboring tissues. Pain syndrome may appear. Blood is detected in the urine, the patient feels a feeling of heaviness and discomfort. Similar symptoms are caused by internal bleeding.
Symptoms
Basically, adrenal myelolipoma occurs without pronounced symptoms and therefore a person may not even be aware of the presence of this pathology in his body over a long period of time, but in some cases hemorrhage occurs into the tumor formed in the adrenal gland. As a result, the patient experiences severe heaviness and pain, the main location of which is the lumbar region. In addition, symptoms such as increased blood pressure and the presence of blood in the urine may also occur. Very often, this pathology is diagnosed completely by accident during an examination of internal organs.
Diagnostic methods
Treatment of myelolipoma begins with an examination by a doctor and analysis of the patient’s complaints. The disease is similar to kidney pathologies of inflammatory origin. You will need to consult a urologist, nephrologist, or oncologist.
The survey plan includes the following activities:
- General urine and blood tests, blood sugar test.
- CT, MRI - the examination method allows you to determine the diameter of the myelolipoma, determine the type and shape, exclude a malignant nature, metastases from other formations, and assess the degree of compression of neighboring organs. It is the main diagnostic tool for lipomatosis.
- X-ray – used if it is impossible to undergo a tomography. When pressure is applied, organs are displaced from their normal location, which can be seen on x-rays. The disadvantage of the method is the inability to estimate the size of the lipoma and determine the type; small tumors are not detected.
- Ultrasound examination is indicated for suspected myelolipoma caused by excess production of male sex hormones in women. Due to the structural features of the body, pathology of the right adrenal gland is easier to diagnose using ultrasound than the left one.
- Biopsy - fine needle or puncture with a thick needle. Indicated in doubtful cases to exclude a malignant nature.
The examination tactics are developed by the doctor depending on the patient’s complaints.
Diagnostic research methods
There are several diagnostic methods that in 90% of cases make it possible to make an accurate diagnosis. The main ones are:
- CT (computed tomography) - determines the density of adenomas in the adrenal glands, which should not exceed the density of adipose tissue (from -25 to -115 NU). Elevated levels may indicate the presence of retroperitoneal sarcoma. Computed tomography allows you to determine the exact localization of the tumor in the walls of the adrenal glands, and not in the kidneys or other organs.
- CT perfusion (CT with contrast) will allow you to more accurately determine the size and characteristics of the tumor. CT with contrast is performed only in cases where the patient does not have allergic reactions to the contrast agent.
- MRI (magnetic resonance imaging) is one of the most accurate research methods. In images taken with an MRI scanner, it is possible to identify tumors of various types.
- Biopsy – performed to detect cancer. The procedure involves taking cells and tissues from the affected organ and helps determine the type of tumor.
- X-ray – allows you to see tumors of sufficiently large size that displace organs from their usual location. This type of examination has low efficiency.
- Ease of examination.
- Quick results.
- The ability to distinguish metastases from benign formations and indicate the site of metastasis.
- Detecting cancer in its early stages.
- Safety of the procedure.
- Laboratory tests - help determine the levels of adrenaline, cortisone, aldosterone.
Cytological examination - establishes an accurate diagnosis of all types of tumors at various stages of development. The advantages of this diagnostic method include:
Adrenal neoplasms in children
Pheochromocytoma and paraganglioma
Tumors of chromophine cells that produce catecholamines. Pheochromocytoma refers to formations of the medullary part of the adrenal glands, tumors of chromophine tissue of extra-adrenal localization, respectively, paragangliomas. Pheochromocytoma and paraganglioma are rare childhood tumors. Among children with arterial hypertension, the incidence of surgically confirmed pheochromocytomas ranges from 0.8-1.7%.
Classification
According to localization, pheochromocytomas are divided: a) into adrenal (90% of cases): bilateral, unilateral; b) extra-adrenal: in the paravertebral sympathetic ganglia; intra- and extraorgan accumulations of chromaffin tissue;
Based on morphology, the following are distinguished: - benign pheochromocytomas; - malignant pheochromocytomas; - multicentric pheochromocytomas (are the result of a total genetic lesion of the adrenal medulla).
According to the clinical course, three forms of pheochromocytoma are distinguished:
- asymptomatic
- paroxysmal; persistent (90% in children); mixed;
- hypotonic form;
Pheochromocytomas are classified according to the severity of their course:
- mild course
- moderate severity
- severe course
Etiology and pathogenesis
Unlike the adult population, children, in most cases, have a clear genetic predisposition to the occurrence of F., with a tendency to bilateral damage to the medullary part of the adrenal glands or extraorgan localization of the formation. Among children, the causes of F. can be a manifestation of several hereditary diseases transmitted in an autosomal dominant manner, including: - multiple endocrine neoplasia (MEN) 2A, or Sipple syndrome (in combination with medullary thyroid cancer (thyroid gland) and hyperplasia and/ or adenomas of the parathyroid glands); — MEN type 2B, or Gorlin syndrome (in combination with medullary thyroid cancer, Marfan-like appearance, multiple ganglioneuromas of the gastrointestinal mucosa (GIT)); - von Recklinghausen's disease (in combination with cutaneous neurofibromatosis); - von Hippel-Lindau syndrome (in combination with retinal hemangiomatosis, spinal hemangiomas and hemangioblastomas, less often with kidney cancer, multiple cysts of the kidneys and pancreas). mutations in the succinate dehydrogenase (SHD) gene and its subunit genes.
Damage to the adrenal glands in MEN 2 syndromes is, as a rule, bilateral in nature; tumors of extra-adrenal localization are usually not detected. In von Hippel-Lindau syndrome, von Recklinghausen disease, and mutations in the SHD genes and its subunits, the tumor from chromaffin cells has both adrenal and extra-adrenal localization.
Clinical manifestations
Clinical manifestations of pheochromocytoma are caused by excess production of catecholamines (adrenaline, normadrenaline, dopamine).
The classic triad of symptoms includes: recurrent headaches, sweating and tachycardia.
Also clinical manifestations of F. are: arterial hypertension in 65% of cases, more often of a paroxysmal nature; up to 30% of children may experience a feeling of discomfort in the back or abdominal cavity, due to pressure of the tumor on adjacent tissues.
Other signs of F. that are less common are pallor, orthostatic hypotension, constipation, mental disorders, blurred vision, weight loss, polyuria, polydipsia, hyperglycemia, and dilated cardiomyopathy.
Diagnostic tactics
Determination of the total concentration of metanephrines (metanephrine and normetanephrine) in plasma and conjugated metanephrines in urine. This is the most reliable method for diagnosing pheochromocytoma. Even with a low level of catecholamines in the blood, the level of metanephrines in the blood is always elevated in pheochromocytoma. Metanephrines are stable for 24 hours, so their determination is not time-related to the moment of hormone release by the tumor. The method has high sensitivity and specificity (reach 98%).
Tumor localization is usually determined using ultrasound (US), computed tomography (CT), and magnetic resonance imaging (MRI). The sensitivity of all methods is quite high, 90-96%.
For accurate topical diagnosis, scintigraphy with meta-123I-benzylguanidine is used. This compound is concentrated in the cells of chromaffin tissue and is integrated during the synthesis of catecholamines. Unmodified adrenal tissue rarely absorbs the isotope, but up to 90% of pheochromocytes absorb it.
Genetic diagnostics
In children, it is important to conduct a gene study: In case of an isolated increase in metanephrines: typing of the RET and MEN2 gene is important, the search for VHL and SHD is not advisable. With an isolated increase in normetanephrines, it is preferable, first of all, to study the VHL and SHD genes. In the case of a mixed type, a study of the entire spectrum of known genes is indicated.
Treatment
Pharmacological preoperative preparation is mandatory. It is carried out to reduce the impact of catecholamine intoxication on target organs: heart, kidneys, brain, as well as to prevent the development of hemodynamic crises during the upcoming operation.
A variety of selective and non-selective blockers of both a- and b-adrenergic receptors with varying durations of action are used. At the initial stage of using a-blockers, significant postural arterial hypotension may be noted.
In the presence of tachycardia, cardioselective ß-blockers are added to therapy with A-blockers. Criteria for the effectiveness of preoperative preparation: Reduction (disappearance) of hypertensive attacks, relief of hypovolemic syndrome, leveling of rhythm disturbances, correction of metabolic disorders.
Adrenocortical cancer
An extremely rare disease among the pediatric population (0-21 years).
Due to the rarity of this pathology, the causes and the possibility of genetic predisposing factors are still not fully understood. Although there are a number of syndromes associated with the development of ACC, including: Lie-Fraumen syndrome, Beckwett-Wedemann syndrome, MEN-1 syndrome, Carney complex, hemihypertrophy syndrome.
According to available data, there is an increase in the incidence of ACC among girls in the age group from 0 to 3 years and after 13 years. There are no population differences in the prevalence of ACR, with the exception of Southern Brazil, where the incidence of ACR is 10-15 times higher than in other populations. This is caused by a mutation in the P53 gene (a tumor suppressor gene). According to the international pediatric registry of ACR, the median development of ACR is 3.2 years, 60% are younger than 4 years and 13% are older than 13 years.
Clinical manifestations.
Unlike adults, ACR in childhood is characterized by hormonal activity in up to 95% of cases. Moreover, in 60% of cases, the tumor produces exclusively androgens, the clinical manifestation of the excess of which is accelerated growth rates, physical development, the appearance of pubic and axillary hair, acne vulgaris, low timbre of voice and signs of gonadotropin-independent premature sexual development of the isosexual type in boys (enlarged external genitalia organs with pre-pubertal testicular sizes) and signs of heterosexual gonadotropin-independent PPR in girls (clitoral hypertrophy). In 30% of cases there is a combination of excess production of androgens and glucocorticoids, and only in 5% of cases glucocorticoids are isolated. Clinical signs of excess glucocorticoids are the syndrome of “hypercortisolism”: dysplastic obesity, stretch marks, hypertrichosis, arterial hypertension, growth retardation.
In 5% of cases, ACC may not exhibit hormonal activity; in this case, the diagnosis is more often established either due to complaints caused by the “mass effect” of the tumor on surrounding organs and tissues, or during a routine ultrasound of the abdominal cavity.
Diagnostic tactics
Laboratory research:
blood sugar, cortisol, ACTH, DHEA-S, 17-hydroxyprogesterone, testosterone. Determination of 24-hour urine for metabolites of metanephrines and normetanephrines. These studies will allow us to establish the presence or absence of hormonal activity of the formation, as well as differentiate between the formation of the adrenal cortex and neuroblastoma, pheochromocytoma.
Instrumental examinations:
An adrenal tumor is easily visualized using an MRI or CT scan of the abdomen. During topical diagnosis, be sure to pay attention to the characteristics of the tumor: a homogeneous hyperechoic structure is more often characteristic of myelolipoma, low density (less than 20), a rapid decrease in signal intensity after contrast is more often characteristic of adenoma or hyperplasia.
If an oncological process is suspected, a CT scan of the chest and a scan of the skeleton bones are also indicated to exclude metastatic damage to organs.
Performing a puncture biopsy for the purpose of differential diagnosis is currently strictly contraindicated in order to prevent metastatic seeding of the body.
Differential diagnosis:
in the presence of a hormonally active formation of the adrenal cortex, differential diagnosis is carried out between ACC, primary adrenal adenoma and secondary adenoma, due to a violation of the hypothalamic-pituitary regulation.
In children under 5 years of age, it is necessary to exclude neuroblastoma, Wilms tumor; in older children, lymphoma, sarcoma, germinal tumors, as well as neuroblastoma.
Forecast
Complete cure is possible with timely complete removal of the formation, especially if the size is less than 9 cm, and the weight of the formation is less than 200 g. In the presence of metastatic damage to other organs, the prognosis is unfavorable. On average, when aggressive surgical treatment is used in combination with chemotherapy, survival is 18 months.
Primary hyperaldosteronism
The disease is caused by increased production of aldosterone by the zona glomerulosa of the adrenal cortex.
It is extremely rare in children.
Among the main reasons are:
- unilateral or bilateral adrenal hyperplasia
- adrenal adenoma
- glucocorticoid-dependent form of hyperaldolateralism.
Clinical manifestations:
because Aldosterone acts on the distal tubules of the kidneys, causing sodium retention and increasing the excretion of potassium and hydrogen ions; hyperaldosteronism is accompanied by hypokalemia, metabolic alkalosis and arterial hypertension. Frequent symptoms of hypokalemia are weakness and polyuria, arterial hypertension.
Diagnostics.
Primary hyperaldosteronism is characterized by an increased level of blood aldosterone and a decreased level of renin or plasma renin activity. Given that each of these indicators varies significantly depending on a number of factors, the most reliable criterion at present is the study of the aldosterone-renin ratio. However, as with other tests, both false positive and false negative results are possible.
Hypokalemia
– can occur in up to 60-70% of cases; its absence does not exclude the presence of PHA.
Topical diagnosis: aldosteromas are most often small in size; CT and/or MRI can be used for their topical diagnosis
In children, especially in cases of lack of accurate topical diagnosis, comparative selective venous blood sampling from the venous vessels of the adrenal glands is indicated in order to lateralize the formation and/or exclude bilateral damage to the adrenal glands
Treatment
The only treatment for aldosteroma is its removal. For bilateral zona glomerular hyperplasia, drug treatment with spironolactone (1-3 mcg/kg/day in 3 doses orally) is preferable, since adrenalectomy usually does not eliminate arterial hypertension. Hyperaldosteronism that responds to glucocorticoid therapy is treated with glucocorticoids in physiological replacement doses.
Neuroblastoma
Neuroblastoma develops from neural crest cells and is localized in the adrenal glands or along the sympathetic trunk. Neuroblastomas in young children are extremely malignant. However, if neuroblastoma occurs in the first year of life, spontaneous regression is often observed. Neuroblastoma is the second most common malignant solid tumor in children (the first is brain tumors) and most often occurs between the ages of 1 and 3 years. At least 50% of these tumors are localized in the abdominal cavity (of which 35% are in the adrenal glands), and 70% of patients already have metastases by the time the diagnosis is made.
Clinical picture
may be erased, or consist of a complex of “small signs”
Frequently indicated symptoms in the literature: Abdominal neoplasm, periorbital edema, weight loss, anorexia, bone pain (metastases), anemia, fever, symptoms caused by excess catecholamines, arterial hypertension, facial flushing, sweating, tachycardia, headache.
Laboratory diagnosis of neuroblastoma is based primarily on determining the content of vanillylmandelic acid (the end product of tissue metabolism of adrenaline and norepinephrine) and homovanillic acid (the end product of dopamine metabolism) in the urine. Usually, during the initial examination, the content of vanillylmandelic acid is determined, but in 15-20% of patients the level of vanillylmandelic acid in the urine is normal, but the content of homovanillic acid is increased. Therefore, both compounds should be determined. Adrenaline levels remain normal.
Mass examinations and prevention. Studies conducted in Japan have shown that detection of neuroblastoma at the preclinical stage according to the determination of vanillylmandelic or homovanillic acid in the urine in children 6 months of age and timely treatment significantly increases survival (up to 97% of patients are cured). The vanillylmandelic acid/homovanillic acid ratio in children at the preclinical stage of neuroblastoma was higher than in children in whom the tumor was diagnosed after the appearance of clinical signs. These data suggest that neuroblastoma cells at the preclinical stage secrete predominantly epinephrine and norepinephrine and are therefore more differentiated.
Another laboratory marker is Cystathionine. Normally, cystathionine is not detected in urine; the presence of cystathionine in urine indicates the presence of neuroblastoma. However, the absence of cystathionine in the urine is not a reason to exclude the diagnosis of neuroblastoma.
The main screening method for diagnosing neuroblastoma is ultrasound, which allows it to be localized, determined its size, and visualized the abdominal organs and retroperitoneal space if the presence of metastases is suspected. The main way to visualize neuroblastoma is computed tomography.
Treatment in the early stages of the process - without signs of metastasis and germination outside the adrenal capsule - adrenalectomy. In case of more severe staging of the disease, chemotherapy and radiation therapy regimens are used along with surgical treatment.
Forecast
The younger the child’s age, the more favorable the prognosis. Survival rate averages 30-35%. To predict the development of the disease and assess the effectiveness of treatment, it is necessary to periodically determine the content of catecholamines in the serum.
It is customary to distinguish between 2 clinical and biological variants of neuroblastoma:
1. Favorable: the tumor is diagnosed at an early age and at an early stage of development. Tumor cells are characterized by triploid karyotypes, the absence of mini-chromosomes, and a low level of amplification (low copy number) of the N-myc oncogene. There are signs of differentiation of tumor cells (predominance of the synthesis of adrenaline and norepinephrine). The prognosis is favorable with minimal or even no treatment.
2. Unfavorable: tumor cells are poorly differentiated; the cell cycle is shortened; the level of amplification of the N-myc oncogene is high. The prognosis is unfavorable. Any methods of therapy (for example, bone marrow transplantation) are ineffective. The first variant of neuroblastoma almost never develops into the second.
Ganglioneuroblastoma.
This tumor contains both neuroblasts and ganglion cells at various stages of differentiation; is formed by transformation of neuroblastoma cells.
Clinical picture
In addition to the symptoms described for neuroblastoma, chronic diarrhea may occur.
Laboratory diagnostics. High concentrations of dopamine, norepinephrine, VIP and prostaglandins may be detected in serum and urine.
Instrumental and radiation diagnostics. 50% of abdominal radiographs show paravertebral calcifications; stretching of intestinal loops is also observed. Ultrasound and CT scans can usually detect a tumor. Thus, the presence of paravertebral calcifications and distension of intestinal loops in a child suffering from chronic diarrhea indicates the presence of a tumor secreting VIP.
The prognosis is more favorable than with neuroblastoma.
The possibility of using laparoscopic adrenalectomy for the treatment of malignant pathology of the adrenal gland is widely discussed in professional circles and has its supporters and opponents. Most specialists - endoscopic surgeons dealing with the problem - agree that in the early stages of the disease, when the tumor does not invade the adrenal capsule and in the absence of metastases, laparoscopic adrenalectomy is a low-traumatic, effective and safe method of surgical treatment, subject to the principles of surgical oncology - ablastics and gentle work with fabrics. Of course, the operation requires a highly qualified operator.
Incidentalomas of the adrenal gland
Tumor-like formations of the adrenal gland are different in their morphological structure, are clinically manifested and do not have the characteristics of malignant growth. They are discovered by chance and are quite rare in childhood. These include benign tumors of the adrenal gland - myelolipoma, ganglioneuroma, adenoma, etc.
Treatment
– surgical as a rule – adrenalectomy.
Adrenal cysts
It is also a rare phenomenon in the pathology of the adrenal gland in children. More often they are of endothelial origin and are single-chamber. Cysts are one of the few neoplasms for which the choice of organ-preserving intervention is appropriate - resection of the cyst, puncture and sclerosis in order to preserve intact adrenal tissue.
Technique of laparoscopic adrenalectomy. Considering in most cases the need to remove the altered adrenal gland from the abdominal cavity using an endobag, it is advisable to use a 10 mm laparoscope. After installing a 10 mm trocar in the navel area, under the control of a laparoscope, 5 mm manipulation trocars are installed in such a way that the lines drawn through the trocar insertion sites and the projection of the adrenal gland form an isosceles triangle, with an apex of more than 90 degrees. The side of the lesion should also be taken into account. The right adrenal is located medial to the left, covered by the lower edge of the liver and has a shorter vein, which should be ligated first in the presence of an actively secreting tumor. The left adrenal gland is in close contact with the spleen, stomach and pancreas. In case of suspected or confirmed malignant process, minimal mechanical trauma to the tumor is one of the basic rules of surgical oncology.
The parietal peritoneum is opened starting from the renal-adrenal fold medially, followed by mobilization of the main venous trunk. In case of technical difficulties in mobilizing the main vascular bundle, it is possible to perform a careful dissection along the periphery of the tumor (adrenal gland). It should be borne in mind that the adrenal gland usually has several paths of arterial blood supply: the main bundle (from the renal artery), the middle adrenal artery (from the aorta) and branches from the inferior phrenic artery. Accordingly, the active use of bipolar coagulation is necessary when mobilizing the superior and medial surfaces of the adrenal gland. The venous branch on the right is short and flows into the inferior vena cava, while on the left it is longer and flows into the renal vein. After ligation of the vascular structures, manipulations must remain careful to avoid the expression of cellular elementals. After mobilization of the altered adrenal gland, the latter is placed in an endobag and evacuated from the abdominal cavity. The hemostasis of the adrenal gland bed is monitored. Some specialists place hemostatic material in the bed of the removed adrenal gland.
Technique for resection of a simple adrenal endothelial cyst. The parietal peritoneum is opened in the area of the renal-adrenal junction. The ventral surface is mobilized away from the peritoneum carefully so as not to damage the thin wall of the cyst. The cyst is visualized. The adrenal gland is mobilized to the extent of clear visualization of unchanged gland tissue. Then the cyst is punctured; the contents must be examined cytologically to confirm the absence of tumor cellular elements. Then, by traction on the cyst shells, using minimally invasive instruments (for example, an ultrasonic scalpel dissector), the shells are completely excised.
In the postoperative period, children are prescribed analgesics and short-term infusion therapy. One or two doses of antiproteolytic drugs (contrical) with monitoring of blood amylase activity can be considered a special feature, taking into account possible intraoperative trauma to the pancreas.
Literature.
1. Al-Otaibi KM.Laparoscopic adrenalectomy: 10 years experience.Urol Ann. 2012 May;4(2):94-7. 2. Amar L, Bertherat J, Baudin E, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. Dec 1 2005;23(34):8812-8. 3. Balassy C, Navarro OM, Daneman A. Adrenal masses in children. Radiol Clin North Am. Jul 2011;49(4):711-27, vi. 4. Bradley CT, Strong VE. Surgical management of adrenal metastases. J Surg Oncol. 2014 Jan;109(1):31-5. 5. Bravo EL, Tagle R. Pheochromocytoma: state of the art and future prospects. Endocr Rev 2003;24:539–553. 6. Chen QL, Su Z, Li YH, Ma HM, Chen HS, Du ML. Clinical characteristics of adrenocortical tumors in children. J Pediatr Endocrinol Metab. 2011;24(7-8):535-41. 7. Conzo G, Pasquali D, Della Pietra C, Napolitano S, Esposito D, Iorio S, De Bellis A, Docimo G, Ferraro F, Santini L, Sinisi A. Laparoscopic adrenal surgery: ten-year experience in a single institution. BMC Surg. 2013;13 Suppl 2:S5. 8. de Barros F, Romão RL, de Pinho-Apezzato ML, Prieto Velhote MC, Schilaich Ricardi LR, Gonçalves Leal AJ, Aoun Tannuri AC, Carvalho B, Odone-Filho V, Tannuri U. Laparoscopic adrenalectomy in children for neuroblastoma: report of case series. Surg Laparosc Endosc Percutan Tech. 2012 Feb;22(1):79-81. 9. Gelas T, Frappaz D, Berlier P, Mouriquand PD, Mure PY.Mini-invasive surgery for adrenocortical carcinoma in children: is it safe?. Pediatric Hematol Oncol. 2013 May;30(4):288-90. 10. Gershuni VM, Bittner JG 4th, Moley JF, Brunt LM. Adrenal myelolipoma: operative indications and outcomes.J Laparoendosc Adv Surg Tech A. 2014 Jan;24(1):8-12. 11. Goldstein RE, O'Neill JA, George W. et al. Clinical experience over 48 years with pheochromocytoma. Ann Surg 1999; 229: 6: 755–759. 12. Gosse P, Tauzin-Fin P, Sesay MB, Sautereau A, Ballanger P. Preparation for surgery of phaeochromocytoma by blockade of alpha-adrenergic receptors with urapidil: what dose?. J Hum Hypertens. Sep 2009;23(9):605-9. 13. Heloury Y, Muthucumaru M, Panabokke G, Cheng W, Kimber C, Leclair MD. Minimally invasive adrenalectomy in children. J Pediatr Surg. 2012 Feb;47(2):415-21 14. International Pediatric Endosurgery Group. IPEG guidelines for the surgical treatment of adrenal masses in children. J Laparoendosc Adv Surg Tech A. Mar 2010;20(2):vii-ix. 15. Kelleher CM, Smithson L, Nguyen LL, Casadiego G, Nasr A, Irwin MS, Gerstle JT. Clinical outcomes in children with adrenal neuroblastoma undergoing open versus laparoscopic adrenalectomy. J Pediatr Surg. 2013 Aug;48(8):1727-32. 16. Laje P, Mattei PA. Laparoscopic adrenalectomy for adrenal tumors in children: a case series. J Laparoendosc Adv Surg Tech A. 2009 Apr;19 Suppl 1:S27-9. 17. Lopes RI, Dénes FT, Bissoli J, Mendonca BB, Srougi M. Laparoscopic adrenalectomy in children. J Pediatr Urol. 2012 Aug;8(4):379-85. 18. Maiti A, Chatterjee S. Congenital adrenal hyperplasia: an Indian experience. J Paediatr Child Health. 2011 Dec;47(12):883-7. 19. Michalkiewicz E, Sandrini R, Figueiredo B, et al. Clinical and outcome characteristics of children with adrenocortical tumors: a report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol. Mar 1 2004;22(5):838-45. 20. Nerli RB, Reddy MN, Guntaka A, Patil S, Hiremath M. Laparoscopic adrenalectomy for adrenal masses in children. J Pediatr Urol. 2011 Apr;7(2):182-6. 21. Neumann HPH et al. Germ-line mutations in nonsyndromic pheochromocytoma. New Engl J Med 2002; 346: 1459–1466. 22. Poddubnyĭ IV, Tolstov KN, Orlova EM, Isaev AA, Gorodnicheva IuM, Oganesian RS Khirurgiia (Mosk). 2011;(9):53-9. Russian. 23. Ribeiro RC, Figueiredo B. Childhood adrenocortical tumors. Eur J Cancer. May 2004;40(8):1117-26. 24. Solaini L1, Arru L2, Merigo G2, Tomasoni M2, Gheza F2, Tiberio GA2. Advanced sealing and dissecting devices in laparoscopic adrenal surgery. JSLS. 2013;17(4):622-6. 25. Sredni ST, Alves VA, Latorre Mdo R, Zerbini MC. Adrenocortical tumors in children and adults: a study of pathological and proliferation features. Pathology. Apr 2003;35(2):130-5. 26. Stewart JN, Flageole H, Kavan P. A surgical approach to adrenocortical tumors in children: the mainstay of treatment. J Pediatr Surg. May 2004;39(5):759-63. 27. Stratakis C, Chrousos G. Endocrine tumors. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. Philadelphia, PA: Lippincott Williams & Wilkins; 1997:947-76. 28. Waguespack SG, Rich T, Grubbs E, et al. A current review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. May 2010;95(5):2023-37. 29. Dedov I.I., Beltsevich D.G., Kuznetsov N.S., Melnichenko G.A. Pheochromocytoma. M: Practical Medicine 2005; 216. 30. Kalinin A.P., Kazantseva I.A., Polyakova G.A. Adrenal and extra-adrenal pheochromocytomas. Tutorial. M 1998; 36.
Forecasts
The prognosis for this pathology is positive. After removal of a tumor in a person, cases of relapse are possible in rare cases.
Treatment of the disease without surgery lasts 5-20 years. The tumor has little effect on the human body. For this reason, patients over 70 years of age do not undergo surgery to remove it.
In rare cases, the neoplasm has an effect on hormonal levels, which can be eliminated by taking appropriate medications and performing radiation therapy.
Life expectancy with adrenal myelolipoma is very high.
Source: UroHelp.guru
Interpretation of results
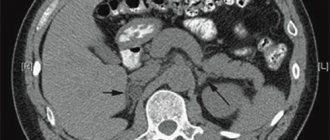
Normally, a CT scan of the adrenal glands reveals the usual location of the glands. The shape of the right adrenal gland is often linear, the left one is V or Y-shaped, the contours are smooth and clear, the density is not changed, the structure is homogeneous, the dimensions are not increased. No changes were detected in the perinephric tissue. Enlarged lymph nodes are not detected at the examined level. After contrasting, both adrenal glands accumulate contrast evenly.
Adenomas detected by CT scan of the adrenal glands are distinguished by their oval or rounded shape, smooth contours, homogeneous or moderately heterogeneous structure. The sensitivity of adrenal CT in detecting adenomas is 98%. With adrenal cysts, CT scan reveals clear, round, thin-walled formations with homogeneous content. The walls of the cysts may have areas of calcification; the density of their contents does not change with contrast. CT visualization of adrenal lipomas is characterized by a rounded shape, clear contours, and moderate heterogeneity of the structure due to fatty inclusions. Lipomas do not deform the adrenal gland and are clearly demarcated from unchanged tissue.
On CT scanning of the adrenal glands, hematomas appear as encapsulated oval formations with uneven contours and a heterogeneous structure due to the presence of clots. Pheochromocytomas detected on CT scanning of the adrenal glands have a heterogeneous structure, round or oval shape, smooth contours, and a well-defined capsule. X-ray absorption increases with contrast. CT signs of malignant tumors of the adrenal glands include significant tumor sizes, heterogeneity of structure with foci of varying density, blurred contours, zones of calcification, enlargement of paravasal lymph nodes, high density of tumor tissue compared to benign findings, and sometimes bilateral localization.
Thanks to the size, contours, shape, changes in density, the presence of inclusions and altered lymph nodes determined by CT scan of the adrenal glands, a specialist can assume the morphological structure of the identified formation and develop further tactics.
Forecast of the course of the disease
The prognosis for adrenal myelolipoma is favorable. For small formations, surgical treatment is not performed. The patient is under dynamic observation. Conservative therapy and diet do not contribute to the regression of the disease. The patient should be informed about the risk of capsule rupture in order to know how to act in this case.
For significant cases, surgical treatment is performed. The result is no compression, no damage to adjacent tissues, and removal of the tumor. The consequences depend on the type, size, and degree of damage to neighboring organs - kidneys, spleen.
After removal of adrenal myelolipoma there is no risk of recurrence. The need for chemotherapy and radiation is not indicated.
If suspicious symptoms appear, contact a nephrologist, urologist and undergo a comprehensive examination. It will allow you to identify the cause of the pathological process and exclude the malignant nature of the tumor. By delaying contacting a doctor, you aggravate the course of the disease.
How to treat adrenal disease?
Nodular diseases of the adrenal glands with surgical indication in most cases are treated using laparoscopic adrenalectomy (Fig. 1) unilateral (functional adenomas, incidentalomas, pheochromocytomas) or bilateral (nodular hyperplasia of the adrenal cortex, Cushing's disease, relapse after pituitary surgery). Traditional open surgery or freezing (cryodestruction) of the adrenal glands is used in rare cases.
During laparoscopic adrenalectomy, the patient receives general anesthesia and is placed in the lateral position (Fig. 2).
In the case of pheochromocytoma, the patient receives intervention with alpha blockers (doxazosin, phenoxebenzomin), beta blockers, calcium channel blockers are also used.
In the case of primary aldosteronism, blood pressure control is achieved by administering an aldosterone antagonist, this is due to the integration of potassium in the case of hypokalemia.
Before surgery, a catheter is placed in the bladder while the patient is asleep. Typically, 4 to 5 small incisions are made to insert laparoscopic instruments. The operation can be performed via a transperitoneal or retroperitoneal approach based on a thorough preoperative examination. In some cases, you can resort to partial removal of the adrenal gland. The average duration of surgery, adrenalectomy, is about 2 hours. At the end, a drainage is installed on the left, which is usually removed on the 2-3 postoperative day. The average postoperative length of hospitalization is 3-4 days. The patient is able to walk, eat on the first day after surgery, and replacement therapy is eventually administered.
Adrenalectomy (laparoscopica.png)
Laparoscopy of the adrenal glands (Fig. 1.)
Laparoscopic adrenalectomy 2.png
Features of kidney treatment
Myelolipoma of the kidney and adrenal gland has a slow growth rate. If the size of the formation is small, dynamic monitoring of the patient's condition is indicated. The duration of this period can be from 5 to 25 years.
- What is angiomyolipoma and its causes?
- Methods of treatment and diagnosis of angiomyolipoma of the right and left kidney
- Features of localization, causes of angiolipoma
For large tumors, compression of neighboring organs, and hematuria, the only treatment option is surgery. The goal is to eliminate tissue compression, normalize blood circulation and general condition.
Indications for surgery:
- suspicion of the onset of a malignant process,
- signs of lipoma growth,
- inflammatory changes in the tissues around the tumor,
- dimensions greater than 60 mm in diameter,
- constriction of the kidney vessels - characteristic of myelolipoma of the left adrenal gland,
- increased blood pressure that is not controlled by taking medications,
- signs of hormonal imbalance associated with education,
- pain, signs of internal bleeding,
- adrenal endocrinopathy.
Surgery is performed laparoscopically. Surgeries in the cavity are prescribed extremely rarely if the malignant nature of myelolipoma is suspected or there are signs of invasion into neighboring tissues.
The use of laparoscopic removal method allows to reduce the length of stay in the clinic. After 3 hours, the patient experiences normalization of intestinal activity and can eat. Discharge is made one day after surgical procedures.
Surgery on the adrenal glands is a complex medical procedure. Related to the location of the organ. The reason for forced refusal of surgical intervention may be the presence of secondary metastases of significant size.
Complications from laparoscopy are unlikely. Secondary infection, swelling, and bleeding are possible. In this case, you should immediately contact a medical facility.
Clinical picture
The occurrence and development of myelolipoma in the human body does not manifest itself with any pronounced symptoms. A person may not realize for many years that he has a benign tumor.
The disease is accidentally discovered during an examination. Health problems are noted only when hemorrhage occurs in the myelolipoma of the endocrine gland itself. A person develops symptoms:
- feeling of heaviness in the lower back;
- stitching pain in the lower back;
- blood pressure surges;
- the presence of traces of blood in the urine when urinating.