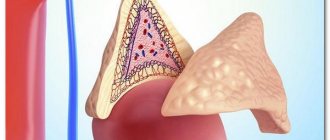
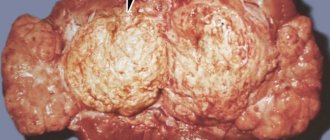
Pheochromocytoma is a rare, usually benign tumor of the adrenal medulla. Only 10% of pheochromocytomas are malignant.
The adrenal glands are paired endocrine glands of a triangular shape, about 1 cm in height and 3.5 cm in length, located in the upper part of both kidneys. Each gland consists of a medulla surrounded by a cortex. The medulla is responsible for producing epinephrine, also known as adrenaline. The adrenal cortex produces other hormones necessary to maintain water-salt balance in the body, such as cortisol and aldosterone. The development of adrenal pheochromocytoma contributes to the release of large amounts of catecholamines into the body, which leads to increased blood pressure and increased heart rate. The development of this disease is possible at any age, but most often it manifests itself between 30 and 50 years of life. Approximately 10% of cases of the disease occur in children. 90% of cases of adrenal pheochromocytoma are sporadic, however, one third of cases may be associated with hereditary causes.
Symptoms of pheochromocytoma
An increased content of catecholamines in the urine can lead to serious disorders of the heart and blood vessels.
In 10% of patients, diagnosis shows a bilateral tumor. In the absence of treatment, deaths were observed due to the development of a hypertensive crisis. In 10% of patients, diagnosis shows a bilateral tumor.
A malignant course poses a great danger. The survival rate of patients is no more than 44% for 5 years after surgery, radiation therapy and taking chemicals.
The production of catecholamines occurs in a pulsating mode, so the symptoms of adrenal pheochromocytoma often appear spontaneous:
During diagnosis, sharp increases in blood pressure occur (from 200).
- sharp increases in blood pressure (from 200);
- increased heart rate;
- anxiety, fear;
- paleness of the skin;
- chest pain.
Signs of pheochromocytoma are acute, the patient develops a stroke or heart attack. The disruption of norepinephrine production is unstable, so the manifestations are constantly changing. This greatly complicates tumor diagnosis.
Against the background of arterial hypertension with pheochromocytoma, the patient experiences headaches and periodically chills. Excessive production of norepinephrine leads to mental disorders of varying severity. There is a disruption in metabolism, the functioning of the stomach and intestines. The lack of oxygen in the brain remains, and leukocytes and sugar increase in the blood.
Recognition
Diagnosis of pheochromocytoma is difficult due to:
- Variability of symptoms;
- Possibility of atypical course of the disease;
- Clinical "masks".
Clinical “masks” are diseases with similar symptoms, which lead to confusion. Pheochromocytoma is often mistaken for thyrotoxic crisis, stroke, myocardial infarction, toxicosis of pregnancy, kidney disease, “acute abdomen”, hypothalamic syndrome and others.
Diagnosis of pheochromocytoma is based on hormonal tests and radiation methods that can visualize the tumor. The purpose of the hormonal stage of diagnosis is to identify increased production of catecholamines. Radiation techniques have the task of confirming or denying the presence of a tumor in the adrenal glands or other areas.
The most accurate indication of a tumor or its absence is the result of the level of metanephrines in the urine per day. This analysis is preferable to detecting the level of catecholamines. This is explained by the fact that pheochromocytoma partially does not release hormones directly into the blood. Metanephrines, the final derivatives of metabolism, enter the blood and subsequently the urine. Determining the amount of metanephrines in the blood is problematic, but a daily urine test is more stable and allows you to record the required value.
Some experts even believe that testing for catecholamines in urine, even 24-hour urine, or blood, is pointless, since there is a high probability of error. The level of these hormones depends on a number of factors that influence the result and can lead to fatal misconceptions about the diagnosis. Having suspected pheochromocytoma, the doctor needs to determine the level of chromogranin A, renin, ACTH, calcitonin, aldosterone, cortisol, and the patient, accordingly, undergo these tests.
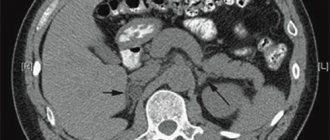
The radiation stage of diagnosing this tumor includes contrast computed tomography. Contrast is injected through the veins; the tinted areas give a clearer picture of the presence or absence of a tumor. The principle is to measure the density of the adrenal gland formation, this is done before the procedure, during and after completion. By comparing data on contrast density before administration, during the study and after its completion, doctors suggest the possible nature of the tumor.
It is believed that tumors that are malignant in nature actively accumulate contrast. Their initial density is slightly higher. The contrast lingers in the tissues for quite a long time. Benign ones are characterized by low density, quickly washed out contrast from tissues, and the density at the final stage is almost identical to the initial one.
In some cases, a metaiodobenzylguanidine scan is used for diagnosis. This is an isotope that malignant tumors actively accumulate in their tissues.
Mechanism of nucleation
Chromaffin cells of pheochromocytoma in the adrenal gland intensively and uncontrollably produce catecholamines. Their constant accumulation leads to the fact that the system of neutralizing the activity of hormones cannot cope with them.
Moreover, over time it only weakens. Therefore, the pathological effect of excess catecholamines on the body systems, the functioning of which they regulate, begins.
In this case, first of all, the functioning of the cardiovascular, nervous and endocrine systems is disrupted.
The main effect of catecholamines is expressed by arterial hypertension. Because hormones, influencing the contractility of the heart, cause rapid heartbeat, vasoconstriction, and a rise in blood pressure.
An equally important negative result of the influence of excess hormones of pheochromocytoma is the acceleration of metabolic processes in the body, which cause disturbances in the functioning of various organs.
- The adrenal glands are not visualized on ultrasound. What does it mean?
When to go to the doctor
A patient who has noticed alarming symptoms indicating the tumor described above should visit an endocrinologist. Of course, the best place to start is by visiting your family doctor, who will refer you to a specialist.
At the beginning of the visit, the specialist will collect a thorough history of the disease and study its nature. It is worth mentioning the moments of exacerbations (before the visit, you need to remember exactly when the first symptoms appeared and how they changed).
It should be kept in mind that symptoms such as:
- change in appetite;
- disturbances in the passage of feces and urine (for example, the need to go to the toilet more often);
- emerging difficulties in falling asleep;
- excessive sleepiness.
A study that confirms that there is excessive production of catecholamines in the body is of primary importance in diagnosing cancer.
Causes
Often, a failure in the production of hormones occurs against the background of the underlying disease for which diagnosis is carried out. The appearance of a formation is one of the indicators of endocrine disorders (thyroid carcinoma, hyperplasia). In 10% of cases, pathologies reveal a hereditary form with autosomal disorders. There is no exact etiology for the development of adrenal tumors.
The appearance of formation is one of the indicators of endocrine disorders caused by an increased content of catecholamines.
The severity of the symptoms does not depend on the shape and size. Even a small formation can seriously affect hormonal levels, increasing catecholamines in the urine and blood. In pathology, calcitonin, serotonin and neuropeptides are excessively secreted. The average size of an adrenal tumor is 5 cm, the weight usually does not exceed 70 grams.
Therapy methods
The main type of treatment for pheochromocytoma is surgery. But before it is carried out, the patient is prescribed drug therapy. This is due to the fact that before surgery the patient needs to relieve the symptoms of a hypertensive crisis.
Features of surgery
During surgery, a total adrenalectomy is performed, with the removal of the affected adrenal gland along with the formation. Since the probability of tumor localization outside the gland and the presence of multiple formations is quite high, surgeons prefer laparotomy access. Sometimes laparoscopic surgery is possible. If multiple endocrine neoplasia is detected, then they resort to removing both adrenal glands. For multiple tumors, there is no uniform standard for their removal. Often, due to the increased risk, resection of formations is carried out in several stages.
Methods of performing the operation.
- Open access. During this operation, an oblique incision is made along the line of the ribs. Recovery after such an intervention is quite long - about several weeks.
- Laparoscopic access. In the area of the abdominal wall, 3 incisions of 1.5 cm in size are made, through which an endoscope with video equipment and surgical instruments are inserted inside. Recovery takes up to 5 days.
- Retroperitoneoscopic surgery. Access to the tumor is gained through the lumbar region, during which the vessels are cut off during the operation, and the formation is removed using special technology.
During any intervention, surgeons monitor hemodynamics. After the operation, blood pressure must decrease. If this does not happen, then this indicates damage to the renal artery or incomplete removal of the tumor. After 8 days after surgery, the patient must again undergo urine and blood tests for catecholamines.
When a pheochromocytoma is detected in pregnant women, doctors first stabilize their blood pressure, then perform an abortion or cesarean section, and only then remove the formation. If the formation turns out to be malignant, then after surgery a course of chemotherapy is prescribed with cytostatic drugs such as Vincristine, Decarbazine and Cyclophosphamide.
Drug therapy
As already mentioned, drug treatment is a preparatory step for surgery. Thus, to normalize blood pressure and stop tachycardia, a- and b-blockers are prescribed, such as:
- Metoprolol;
- Propranotol;
- Tropaphen;
- Phentolamine;
- Phenoxybenzamine.
Metoprolol is a drug for the treatment of pheochromocytoma.
To reduce the intensity of heart contractions and lower blood pressure, calcium channel blockers (Nifedipine) can also be prescribed, and an inhibitor of catecholamine production (Metyrosine) can be prescribed to suppress the synthesis of adrenaline and norepinephrine.
In another vein, conservative treatment is not used for pheochromocytoma, since it is not effective. Medicines can only lower the level of catecholamines in the body, but do not affect the formation itself. Drug therapy is aimed mainly at stopping the hypertensive crisis. Taking adrenergic blockers for too long can cause mental disorders and abnormalities in the gastrointestinal tract.
Causes of pheochromocytoma
An adrenal tumor is a formation about 5 cm in diameter, its average weight is 70-75 g. It is formed from functioning tissue and is capable of producing hormones. Most cases of the disease are of unknown origin. The most likely factor is a hereditary factor.
Pheochromocytoma is often combined with multiple lesions of the endocrine glands, vascular tumors, thyroid cancer, neoplasms of the intestines, kidneys, and organs of vision.
If there are no cases of pheochromocytoma in the family, then it is considered a sporadic (random) gene mutation. Such tumors are single, usually located only in one adrenal gland, and in hereditary diseases they are found in multiple and bilateral forms.
We recommend reading the article about adrenal diseases. From it you will learn about what diseases of the adrenal glands can be, their hyperfunction and hypofunction, the causes of adrenal diseases, as well as symptoms in children, women and men. And here is more information about hypothyroidism and folk remedies.
Theoretical information
Not everyone knows what pheochromocytoma is, since it is a fairly rare disease that occurs in two patients out of 1 million people. On average, the age of patients is in the range of 20-50 years, with a tenth of them being children. Most often, in childhood, the disease is diagnosed in boys, although in adults the fairer sex is more susceptible to it.
The adrenal glands are paired glands that are designed for internal secretion. The organs are located in the upper part of the kidneys, and each of them weighs 4 grams. The adrenal glands perform a very important function in the human body - the production of hormones that are designed to regulate metabolic processes, and they can also speed up and facilitate the body’s adaptation to negative environmental conditions. The adrenal glands consist of two layers:
- medulla - the inner cavity of the adrenal gland;
- cortex – outer surface.
The adrenal glands are paired glands that are designed for internal secretion
If we look at pheochromocytoma in more detail, we can determine that it is a neoplasm with remarkably well-developed blood circulation, and it is also surrounded by a special capsule. The size of the tumor in each patient varies over a fairly wide range, but the minimum value is 0.5 cm and the maximum is 14 cm. Every year its size will grow by 3-7 millimeters, which is a fairly large indicator. In 9 out of 10 patients, the tumor is benign; however, in the tenth patient, the prognosis will be less favorable, since the tumor will consist of malignant cells.
The tumor can be localized in the area around the adrenal gland or directly affect the gland. If the tumor is located outside the adrenal gland, then only norepinephrine can be produced. In this case, the symptoms of the disease will not be so pronounced, because this hormone itself does not have a pathological effect on the body. As a rule, a formation is diagnosed on one of the adrenal glands, and in 10% two glands are affected at once.
Establishing diagnosis
Pheochromocytoma is diagnosed only after characteristic manifestations - a stable or sharp increase in blood pressure. There are several methods to determine damage to the adrenal gland.
Urine is examined for catecholamines and metanephrines.
- Laboratory diagnosis of pheochromocytoma. A special high-precision test is used to check the composition of daily urine. Urine is examined for catecholamines and metanephrines. A high level of hormones occurs with an atypical tumor of the adrenal gland, for example, paraganglioma of the bladder. This disease must be excluded. Diagnostics do not always detect formation when determining catecholamines. Hormone levels can vary greatly depending on external factors. During the examination, the emphasis is on the amount of chromogranin A. This universal transport protein is produced by all endocrine tumors. An increase in hormones further confirms the diagnosis.
Right adrenal pheochromocytoma on radiological diagnostics.
- Radiation diagnostics. The method involves the use of a CT scanner. A contrast agent is injected intravenously. Pheochromocytoma is easily detected because it accumulates staining contrast. At all stages of radiation diagnostics, tissue density is checked. The most informative is the delayed density, when the contrast is finished injecting. With a benign tumor, the screen shows low density, the contrast quickly accumulates and is washed out of the tissue.
Prevention
In the case of genetic tumors of the adrenal glands, the care of all family members with whom the patient comes is very important. If possible, genetic testing should be performed on immediate family members and children to rule out the presence of mutations responsible for the disease.
If a mutation is detected or this disease syndrome is diagnosed, regular testing of concentrations in human urine is necessary.
This makes it possible to quickly detect a developing tumor. This prevents the development of life-threatening complications and allows the implementation of effective treatment.
Symptoms of the disease
The main manifestation is high blood pressure. It can be stably elevated (up to 160/95 mm Hg) or have the character of a hypertensive crisis. The systolic reading during an attack can exceed 250 mmHg. Art. A jump in blood pressure is provoked by:
- What does an ultrasound scan of the adrenal glands show and how to properly prepare for the study?
- stressful situations;
- physical exercise;
- deep palpation of the abdomen;
- massage;
- straining when emptying the intestines or bladder (if there is a tumor in it);
- exposure to high or low ambient temperatures;
- use of medications.
High blood pressure in pheochromocytoma
The heart muscle is primarily affected by high blood pressure and the stimulating effect of catecholamines. This is manifested by constant and paroxysmal tachycardia, pain in the heart like angina pectoris, and increased heartbeat. If the tumor persists for a long time, catecholamine cardiomyopathy with circulatory failure develops. Complications of the crisis include: myocardial infarction, pulmonary edema, cardiac asthma, aortic aneurysm.
During an attack (sometimes outside of it), patients are concerned about:
- headache,
- dyspnea,
- sweating,
- nausea and vomiting,
- abdominal pain,
- fainting states,
- seizures,
- visual impairment,
- numbness and trembling of the limbs,
- constipation,
- inexplicable anxiety
- fear of death,
- weight loss.
A hypertensive crisis due to an adrenal tumor has a sudden onset and end. Its duration varies from a minute to a day or more. At the end, the pressure drops, profuse sweat is released, and profuse urination is noted . Patients feel completely powerless. The outcome of the crisis is renal, cardiac, vascular failure, and cerebral hemorrhage.
The most severe consequence is catecholamine shock. When it occurs, the pressure changes sharply from hypertension to hypotension, and medications cannot stabilize the levels, which threatens the death of the patient. Malignant tumors cause constant abdominal pain, severe weight loss and early metastasis (spread) to internal organs.
Complications of pheochromocytoma
Constant production of hormones increases blood pressure and can damage the heart, kidneys, and brain. Life-threatening conditions may occur, such as:
- arrhythmia
- myocardial infarction
- stroke
- renal failure
- acute respiratory distress syndrome.
Catecholamine shock
This complication is due to the fact that catecholamines are not inactivated and continue to act on the blood vessels. In this case, the change in pressure becomes unpredictable, episodes of increased pressure are chaotically replaced by hypotension. Alpha-blockers are used for treatment; in severe cases, hospitalization in the intensive care unit is necessary.
Malignant pheochromocytoma
Rarely, malignancy may occur, i.e. malignancy of education. This condition is called malignant pheochromocytoma. The diagnosis is confirmed by a biopsy, detection of metastases in the bones or other organs.
Treatment and preparation for it
With pheochromocytoma, symptoms, diagnosis and timely treatment are closely related.
A radical way to eliminate pheochromocytoma is surgery.
A radical way to eliminate pheochromocytoma is surgery. After tumor removal, the patient's quality of life improves significantly. In 75% of therapy, blood pressure stabilizes, which eliminates the development of complications in the form of heart attacks or hypertrophy of cardiac tissue. Careful preparation of the patient is required before surgery. Severe hypertensive crises are a cause of danger during surgical procedures.
Conservative therapy
The main drug for temporary drug treatment is Doxazosin.
Temporary drug treatment for pheochromocytoma is often used. It includes normalizing blood pressure and eliminating its strong fluctuations. The main drug is Doxazosin. The patient must drink it no later than 14 days before surgery. Then additional diagnostics are carried out. Thanks to the adrenergic blocker, the receptors become insensitive to the release of catecholamines.
- Adrenal hyperfunction: causes, symptoms, diagnosis and treatment
Surgery
Postoperative suture.
With proper preparation of the patient, surgical intervention proceeds without circulatory disturbance. During the removal of a pheochromocytoma, most of the time is spent on accessing the adrenal gland. The surgeon cuts the muscles of the abdominal wall and chest. The average time for surgery is 2 hours.
Treatment of pheochromocytoma
The only effective treatment for chromaffinoma is its radical removal. The operation is performed in two ways: open or laparoscopic.
The open method has a number of advantages: it is accessible, does not require special equipment and skills from the surgeon, allows a good examination of the surgical area, thorough hemostasis, and it is possible to remove tumors of any size. The disadvantages are high trauma, risk of infection, heavy blood loss, long recovery period, cosmetic defect.
The laparoscopic method of removal is the most popular method today. However, it requires certain preparation and material equipment. It is also not applicable for malignant, bilateral and large tumors.
Access points for laparoscopic removal of the adrenal gland. After the operation there is no cosmetic defect, because the size of the wounds does not exceed 2 cm.
The method of performing the operation is determined by the surgeon.
Careful preoperative preparation aimed at stabilizing hemodynamics is important for the successful outcome of the operation. For this purpose, α-blockers (Prazosin, Cardura) are often used. These drugs are also used to relieve hypertensive crisis.
Causes of attacks
When diagnosed with pheochromocytoma, symptoms (diagnosis, by the way, is also possible based on the primary signs of the disease, since they are quite specific) can arise spontaneously. However, they are often caused by certain circumstances, for example:
- physical exercise;
- anxiety or stress;
- change in body position;
- active bowel function;
- prenatal condition and childbirth.
An attack of symptoms can also be triggered by eating foods rich in tyramine, a substance that affects blood pressure. Levels of this component are very high in fermented, pickled, canned, overripe and spoiled foods. So, you should avoid using:
- some types of cheese;
- some brands of wine and types of beer;
- dried or smoked meat;
- avocados and bananas;
- salted fish;
- sauerkraut or kimchi.
There are also medications that you should not take if you have pheochromocytoma. Symptoms that will be diagnosed by a doctor can be caused by the following medications:
- decongestants and decongestants;
- monoamine oxidase inhibitors;
- stimulants (such as amphetamines).
Signs and symptoms of a benign adrenal tumor often indicate the presence of a pathology, but in some cases similar manifestations are characteristic of other, more common diseases. That is why the role of timely diagnosis cannot be underestimated.
Although high blood pressure is recognized as the primary symptom of pheochromocytoma, most people with hypertension do not have adrenal masses. However, it is strongly recommended that you see a GP if any of these factors apply to you:
- Blood pressure is difficult to control with the current treatment plan.
- There is a family history of cases of pheochromocytoma.
- There was a family history of similar genetic disorders: multiple endocrine neoplasia, cerebroretinal angiomatosis (Hippel-Lindau disease), hereditary paraganglioma or neurofibromatosis type 1 (Recklinghausen disease).
Epidemiology of pheochromocytoma
The prevalence of pheochromocytomas is not precisely known, but the incidence may be significantly higher in some populations. The incidence rate is estimated at 2 to 8 cases per 1,000,000 people per year, approximately the same among men and women. There is an increase in the prevalence of pheochromocytomas with age; more than 50% are diagnosed at autopsies, reaching 0.1% in individuals without a lifetime diagnosis. According to the organization that studies pheochromocytomas, PRESSOR, the total incidence is 10 cases per 1,000,000 people per year. Among randomly discovered adrenal tumors, pheochromocytomas turn out to be from 1.5 to 11%, with an average of 5%.
Numerous historical literature points to the “rule of tens,” which states that 10% of pheochromocytomas are extra-adrenal (10% of them are located outside the abdominal cavity), 10% are malignant, 10% are bilateral, 10% are found in patients without hypertension, 10% are found in children and 10% are caused by hereditary diseases. Recent studies have refuted this rule. Up to 98% of tumors are localized in the abdominal cavity and pelvis, less than 2% in the chest and about 0.2% in the neck; extremely rarely they occur in the skull, middle ear and spermatic cord. The proportion of malignant forms of pheochromocytomas is 13-26%.
Causes of disease development and provoking factors
The mechanisms of development of pheochromocytoma of extra-adrenal localization and directly of the adrenal glands are still not fully understood.
When considering the etiology of a disease, there are usually two key components:
- In 10% of subjects, this pathology is a hereditary disease. Its nature is caused by genetic abnormalities and is inherited in an autosomal dominant manner.
- Pheochromocytoma as part of the MEN syndrome type IIa and IIb. In addition, MEN type IIa (Sipple syndrome) includes medullary thyroid cancer and hyperparathyroidism. MEN type IIb differs from IIa in the more malignant course of thyroid carcinoma and the appearance of skeletal deformities.
Provoking factors for a crisis can be emotional or physical stress, exposure to ionizing radiation, smoking and drinking alcohol in large doses.
Pheochromocytoma - what is it?
Pheochromocytoma (also called chromaffinoma) is a hormone-producing tumor that arises from chromaffin cells of the adrenal medulla.
Chromaffin, or pheochromic, cells are located at number 1 and are the main cells of the adrenal medulla
Important Despite the fact that most pheochromic cells have a typical location (adrenal glands), some of them are part of the nerve ganglia and are found in the abdominal aorta, the hilum of the liver and kidney, the bladder, myocardium, mediastinum and many other internal organs.
With regard to the characteristics of the course of pheochromocytoma, the ten percent rule applies:
- Taking into account the peculiarities of the anatomical location of the cells, this tumor can be located either in the adrenal glands (up to 90%), or have any other location and be called paraganglioma (10%).
- According to its properties, pheochromocytoma is a benign tumor; only 10% have signs of malignancy.
- Chromaffinoma most often affects one adrenal gland, but 10% have bilateral localization.
- There is information about the hereditary nature of this disease, but this has been reliably proven in 10% of cases.
The appearance of adrenal pheochromocytomas is varied, but they have some similarities in the anatomical structure. Any chromaffinoma is covered with a capsule that separates it from surrounding tissues and is highly vascularized. Sizes from a few millimeters to 15 centimeters or more.
Important! When studying the hormone-synthesizing ability of a tumor, it was proven that there is no connection between its size and the amount of hormones produced.
The pathogenesis of the disease is based on the appearance of excessive levels of catecholamines in the blood due to the work of atypical cells that do not obey inhibitory mechanisms.
Symptoms
Most patients with pheochromocytoma typically exhibit three classic symptoms:
- headache;
- sweating;
- rapid heartbeat combined with hypertension.
The patient may also have:
- anxiety;
- nausea;
- weakness;
- abdominal pain;
- weight loss.
Some people never develop symptoms of FCC. 10% of cases are discovered incidentally, meaning that they go unnoticed and are only discovered when the patient undergoes diagnostic tests for other illnesses.
Pheochromocytomas are present in only 0.2% of all people with high blood pressure.
Paroxysmal form of the disease
Research shows that 85 out of a hundred patients suffer from the paroxysmal form. In this case, the patient experiences typical signs of a hypertensive crisis:
- My head hurts very badly. Sometimes the pain is accompanied by dizziness.
- A person cannot move normally, as any effort is accompanied by shortness of breath.
- Symptoms also include pain in the heart area.
- The person is restless, pale and subject to an inexplicable feeling of fear.
- With this diagnosis, sweating increases and the number of heartbeats increases.
- Excessive urination occurs.
- Nausea sets in and convulsive syndrome appears. The person also feels feverish and thirsty.
Many of the above symptoms may not occur at the same time, but there are three signs that almost 100% of patients complain about: headache, increased sweating and rapid pulse.
As the name suggests, symptoms of this type of pheochromocytoma are not present all the time. The attack appears suddenly and can be provoked by anything: a person can overdo it with physical activity, eat junk food, drink an alcoholic drink, get nervous, or even just urinate unsuccessfully. A sharp rise in pressure is likely to await anyone who is trying to find an adrenal tumor.
The attack, if you're lucky, can go away in a couple of minutes, but in difficult cases there are often crises lasting several hours. An unexpected end to an attack is very typical for the disease. In this case, the patient may leak up to five liters of urine, and he himself will be literally wet with sweat. The patient experiences weakness and a feeling of incredible fatigue.
Medicines
Most likely, the doctor will prescribe two different medications, which will need to be taken in courses of 7-10 days in order to lower blood pressure before surgery.
Alpha blockers interfere with the primary function of norepinephrine and prevent muscle stimulation in the walls of small arteries and veins. Because these blood vessels remain open and relaxed, blood flow improves and blood pressure decreases. Side effects include:
- visual impairment;
- irregular heartbeat;
- dizziness;
- fatigue;
- swelling of the limbs;
- sexual dysfunction in men.
Beta blockers are prescribed to inhibit the effect of adrenaline. As a result, the heart beats slower and less intensely. In addition, beta blockers keep blood vessels open and relaxed by slowing down the kidneys' production of a certain enzyme.
Side effects may include:
- feeling tired;
- stomach upsets;
- headache;
- dizziness;
- constipation;
- diarrhea;
- irregular heartbeat;
- difficulty breathing;
- swelling of the limbs.
When diagnosed with pheochromocytoma, symptoms that were treated too late develop into complications. That is why doctors prescribe such drugs, regardless of the nature and intensity of potential side effects.
Other drugs that lower blood pressure are prescribed in cases where alpha and beta blockers are unable to stabilize hypertension.
Diagnostics
Adrenal hormones and their functions + table
The following research methods exist:
- laboratory;
- hardware.
Laboratory research methods include the following tests:
- general blood analysis;
- blood test to determine catecholamines.
It must be taken into account that all hormone levels increase tenfold during an ongoing attack.
- Analysis of daily urine sample;
- Analysis of a three-hour urine sample.
It is typical for the course of this disease that outside of an attack, all laboratory parameters return to normal.
CT allows the diagnosis of pheochromocytoma to be made with great accuracy
Hardware research methods:
- Ultrasound - allows you to determine the size of the tumor and its location, the accuracy of the study is 80 - 90%;
- CT is an x-ray research method, layer-by-layer image of an organ. Accuracy – 100%;
- MRI is a non-X-ray research method, with this method you can detect the smallest tumor size. Accuracy – 100%.
- fine-needle puncture – allows you to take a tumor biopsy under ultrasound and CT control;
- topical diagnostics - scintigraphy with a radioactive substance.
Imaging studies
If laboratory tests indicate that you may have a paraganglioma or pheochromocytoma, the symptoms, reviews in many printed periodicals, and laboratory findings will be the basis for ordering imaging tests to determine the location of the tumor. Such studies include:
- computed tomography (CT);
- magnetic resonance imaging (MRI);
- scanning using the injection of a special radioactive substance - metaiodobenzylguanidine;
- positron emission tomography (PET).
Diagnosis of pheochromocytoma
In the diagnosis of pheochromocytomas and paragangliomas, it is important to collect an anamnesis to identify the characteristics of episodes of increased pressure or their clinical equivalents, complications of pheochromocytoma, and identify a family history of the disease. Despite its rarity, physicians should be wary of pheochromocytoma, as untreated disease can be fatal.
When palpating the abdomen, only about 10-20% of tumors are detected. Palpation of the abdomen when searching for pheochromocytoma should be carried out carefully, with preliminary administration of phentolamine intravenously.
Laboratory diagnostic methods
The pathways for the synthesis of catecholamines in the body have been known since 1939. The content of catecholamines in various biological fluids and tissues was assessed using colorimetric and biological sampling methods. Since unchanged catecholamines represent <2% of excreted urine products, diagnostic methods for the determination of various catecholamine metabolites began to develop in accordance with the pathways of catecholamine metabolism: vanillylmandelic acid, metanephrines, as well as homovanillic acid, methoxyhydroxyphenylglycol and other metabolites.
Catecholamines circulate in plasma both in free and bound (in the form of sulfates) form; the biological effect is due to the free fraction. In patients with pheochromocytoma, both fractions are elevated, but measurement of the free fraction is preferable due to the effect of diet on the level of bound catecholamines. Norepinephrine is the main free catecholamine in the blood plasma and represents only a small part of the neurotransmitter that has not been reuptaken by the presynaptic membrane of postganglionic sympathetic neurons. The content of adrenaline in blood plasma is an order of magnitude lower than norepinephrine. Their ratio plays a role in the diagnosis of pheochromocytoma. Plasma dopamine predominantly circulates in bound form, but little is known about its clinical and physiological significance.
The source of norepinephrine and urinary epinephrine are the cells of the adrenergic system, while dopamine is formed primarily as a result of the conversion of 3,4-dihydroxyphenylalanine (DOPA) in the kidney. Dopamine is the main unbound catecholamine in urine; its determination, together with norepinephrine and epinephrine, is important to determine the malignant nature of the tumor.
Vanillylmandelic acid is formed from adrenaline and, to a greater extent, norepinephrine with the participation of the enzymes catechol-O-methyltransferase and monoamine oxidase, and is excreted by the kidneys mainly in free form.
Normetanephrine and metanephrine are metabolites of norepinephrine and epinephrine, respectively, they are formed by the enzyme catechol-O-methyltransferase without deamination.
Based on the position of evidence-based medicine, the first international symposium on pheochromocytomas for the diagnosis of pheochromocytoma and paraganglioma proposes the measurement of catecholamine metabolites in urine or blood as the method of choice. It has now been established that catecholamines are metabolized into metanephrines in tumor chromaffin cells. The metabolic rate does not depend on the release of catecholamines, but depends on the content of the enzyme catechol-O-methyltransferase. Tumors are described that do not themselves secrete catecholamines, but convert them into metanephrines. This phenomenon explains the lesser dependence of the level of metanephrines on the influence of external factors, as well as the fact that the level of metanephrines, and not blood catecholamines, clearly correlates with the size of the tumor. According to some data, knowing the level of plasma metanephrines, one can predict the size of the tumor with an accuracy of up to 30%, as well as predict its location.
Blood sampling for analysis should be done with the patient lying down. To avoid stress in the patient associated with venipuncture, it is recommended to draw blood through a previously placed intravenous catheter.
When assessing the result of an analysis, one should not be guided only by whether it falls within the norm, but the degree of improvement in indicators should also be taken into account. With a slight increase in the level of metanephrines, the diagnosis of pheochromocytoma is doubtful, and with a fourfold increase, pheochromocytoma is detected with a probability close to 100%. In children, indicators are more difficult to assess without using age-specific reference values. However, according to other authors, blood sampling in children can be a stressful event, but the result is easier to assess without using age-specific reference values.
In patients with renal failure, bound metanephrines may be significantly elevated as they are excreted by the kidneys.
When measuring normetanephrine and plasma metanephrine, there is a risk of missing rare tumors that secrete predominantly dopamine, and these tumors usually do not have classic symptoms. To confirm the diagnosis in such patients, it can be recommended to determine dopamine or its metabolite, methoxytyramine.
A single analysis outside of a crisis does not exclude pheochromocytoma, since these tumors can release hormones paroxysmally. Due to the fact that the method may in some cases be uninformative, in the literature this method is figuratively called “Russian roulette”.
Of the suppressive tests, oral Clonidine is the most popular because it causes less blood pressure response but has high sensitivity with low specificity. In healthy people, clonidine, acting on central a2 receptors, inhibits the activity of peripheral sympathetic nerves and thereby reduces the content of catecholamines.
Many authors point out the need for additional research when pheochromocytoma is detected to exclude the development of a tumor as part of hereditary syndromes. These tests include parathyroid hormone testing along with calcium levels, serum calcitonin testing, screening tests for genetic abnormalities, consultation with an ophthalmologist to exclude retinal angiomas, MRI of the brain is recommended to exclude cerebellar hemangioblastomas (Hippel-Lindau syndrome), and CT kidneys and pancreas to exclude cysts.
In recent years, the arsenal of diagnostic tools for pheochromocytoma and other catecholamine-secreting tumors has been replenished with methods such as the determination of thyroxine hydroxylase, neuron-specific enolase, chromogranins A and B, neuropeptide Y, secretogranin, synaptophysin, protein S-100 and other substances. However, along with positive reviews, there are reports of insufficient sensitivity and specificity of the above tests, especially when the excretory function of the kidneys decreases, even to the smallest extent.
Ultrasonography
In large tumors, necrosis cavities with liquid contents and calcification are possible; in their presence, the homogeneous structure is lost. The sensitivity of the method is estimated as 83-89%, i.e. relatively low. When detecting a tumor, ultrasound should be supplemented with CT or MRI. The advantage of the method is that there is no radiation exposure, so the method can be used in certain categories of patients, such as pregnant women and children.
CT scan
CT examination is often used to diagnose pheochromocytomas; the resolution of CT allows one to detect pheochromocytomas from 0.5 cm in diameter. When performing a CT scan, if there are doubts about the interpretation of the data obtained, it is possible to administer contrast, followed by a repeat examination to clarify additional properties of the tumor. Small tumors are usually homogeneous and have a density of 40-50 units. Hounsfield, evenly capture the contrast agent. Cases of pheochromocytomas have been described when their x-ray density was less than 10 units. Hounsfield and they quickly drew a contrast. In this case, there is a high probability of interpreting radiological data with an erroneous diagnosis of adrenal adenoma. According to our data, approximately 11% of tumors had a homogeneous structure similar to the structure of the vast majority of adrenal adenomas. In this regard, the analysis of densitometric indicators of pheochromocytomas revealed an important differential diagnostic criterion, namely a high density index. This indicator averaged 25.38±8.69 HU; it increased significantly after the administration of a contrast agent. Pheochromocytomas usually had clear (84.2%) but uneven (78.9%) contours. Most tumors (73.7%) were surrounded by a thin capsule.
Currently, data on the effect of contrast agents on catecholamine levels are not confirmed.
If a pheochromocytoma/paraganglioma is detected on a CT study using a contrast agent, there is no need for an MRI study, however, scintigraphy can be recommended to confirm the diagnosis and exclude metastases. The sensitivity of the method for detecting pheochromocytomas is 85-94%, paragangliomas - more than 90%. When using a contrast agent, the sensitivity is 98% and the specificity is 92%. Postoperative tissue changes and surgical clips can reduce these indicators.
Magnetic resonance imaging
MRI is the best method for diagnosing pheochromocytomas. On T1-weighted images, pheochromocytoma is defined by its density as the liver and kidneys and can be easily distinguished from adenoma containing adipose tissue. Hypervascularization of tumor tissue appears as an intense signal on T2-weighted images. The same signal, however, can also manifest itself in hemorrhages, hematomas, adenomas and carcinomas.
The advantages of MRI include the high sensitivity of the method (93-100%) and the absence of ionizing radiation; there is also the possibility of using a contrast agent; the disadvantages include the high cost of the study.
Positron emission tomography
The use of positron emission tomography is limited due to the high cost and expensive equipment. Nevertheless, the possibilities of PET for diagnosing pheochromocytomas are being actively studied and are used in some cases. Most often, 2-fluoro-2-deoxy-O-glucose is used to diagnose pheochromocytomas. There is an assumption that the uptake of this substance can detect malignant pheochromocytomas, but since glucose is taken up by all tissues with accelerated metabolism, this indicator for pheochromocytoma remains nonspecific. PET using a labeled glucose derivative is useful for localizing rapidly growing or undifferentiated tumors, as well as for tumors that do not take up metaiodobenzylguanidine.
PET with other substances is also used to diagnose pheochromocytomas. Hydroxyephedrine and adrenaline labeled with 11C are used. The short half-life and high cost significantly limit the use of these substances. PET studies using 18P-DOPA and 18P-dopamine may be promising.
Differential diagnosis
Many authors use the term "pseudopheochromocytoma" to designate a characteristic clinical situation, provided that pheochromocytoma has been excluded. Pseudopheochromocytomas are a heterogeneous group of diseases. Primary pseudopheochromocytomas constitute severe forms of paroxysmal essential hypertension. Secondary are diseases of the adrenal glands, thyrotoxicosis, hypoglycemia, stroke, migraine, epilepsy, myocardial infarction, arrhythmias, panic disorder, “cheese” syndrome, the influence of various pharmacological agents (cocaine, LSD, amphetamine, thyroxine, MAO inhibitors, tricyclic antidepressants) and others causes. For differential diagnosis, it is very important to collect anamnesis and identify the characteristics of the course of paroxysm, as well as identify the conditions under which it occurs. The most difficult is the differential diagnosis with paroxysmal essential hypertension, in which paroxysms occur spontaneously, as with pheochromocytoma.
Manifestations of symptoms
The main symptom of pheochromacytoma is arterial hypertension. In this case, the pressure can be steadily elevated or rise and fall in fits and starts. Such conditions are accompanied by various symptoms. Therefore, adrenal pheochromocytomas are classified according to symptomatic manifestations:
- paroxysmal;
- persistent (constant);
- mixed.
In 85% of diagnosed cases, paroxysmal disease is detected. Patients with this form of pathology suffer from periodic hypertensive crises, which manifest themselves:
- a sharp increase in pressure;
- causeless feeling of anxiety, restlessness;
- trembling in the body, chills;
- redness and swelling of the face;
- profuse cold sweat;
- heart pain, arrhythmia, tachycardia;
- visual impairment (decrease in visual acuity, sparks or spots before the eyes);
- sharp throbbing headache;
- attacks of nausea and vomiting.
In each individual case, the list of symptoms is different, some may be absent. The crisis lasts several minutes or hours. It stops as suddenly as it began, and the pressure drops sharply to normal. The patient becomes weak and experiences excessive urination. A severe attack can be complicated by a stroke, heart attack, or pulmonary edema.
The persistent form of pheochromocytoma is characterized by persistently elevated blood pressure. This type of tumor is rare. In this case it is observed:
- stable hypertension;
- weakness, fatigue;
- emotional instability;
- headache.
Impairments in kidney function, changes in the fundus of the eye, and enlargement of the heart are also possible.
The symptoms of this form resemble various types of hypertension, sometimes thyrotoxicosis. Constant blood pressure holds excess dopamine.
The mixed form of the neoplasm is expressed by arterial hypertension and hypertensive crises after exposure to provoking factors. The attack ends with a decrease in pressure to the initial levels.
The latent course of the disease without symptoms is dangerous. A provoked hypertensive crisis with this form of tumor can be fatal.
Patients with pheochromocytoma lose weight due to metabolic disorders, they develop intestinal atony, and experience pain in the lumbar region. High blood sugar can trigger diabetes.
Neurasthenia often manifests itself. Therefore, an identified and removed tumor will help normalize blood pressure and eliminate other symptoms.
General information about pathology
Pheochromocytomas are classified as endocrine tumors consisting of pheochromic cells that synthesize excessive quantities of adrenal hormones - adrenaline, dopamine and norepinephrine. Such substances are also called catecholamines. Hormonal hyperfunction leads to an increase in blood pressure, provokes vegetative-vascular disorders and metamorphoses in the myocardium and kidneys. As a rule, hypertensive crisis with pheochromocytoma is common.
Pheochromocytoma tumor is often diagnosed in both men and women aged 20 to 40 years. If we talk about children, education is more often found in boys (in 60% of cases). These formations can be either benign or malignant. The latter are diagnosed in less than 10% of cases and are mainly characterized by an extrarenal location. They also often synthesize dopamine. Metastasis of malignant pheochromocytomas occurs in regional lymph nodes, muscles, lungs, liver and bones.
Cancerous (malignant) tumors
In rare cases, pheochromocytoma, the symptoms of which have remained without proper medical attention for a long time, becomes malignant, and cancer cells spread to other parts of the body, forming metastases. The abnormal cells that first develop in a pheochromocytoma or paraganglioma travel to the lymph nodes, bones, liver, or lungs. It is in these organs that secondary tumor foci most often form.
Sources used:
- https://kistateka.ru/pochki/feohromotsitoma-nadpochechnika
- https://urohelp.guru/nadpochechniki/feoxromocitoma.html
- https://endokrinolog.online/feohromocitoma-lechenie/
- https://www.syl.ru/article/200607/new_feohromotsitoma-simptomyi-diagnostika-analizyi-lechenie-k-kakomu-spetsialistu-obratitsya
- https://propochki.info/nadpochechniki/feohromocitoma-nadpochechnika
- https://pochkam.ru/nadpochechniki/feohromotsitoma-nadpochechnika.html